奉贤区新型催化剂及配体研究进展(33)
自1865年Kekulé提出苯的合理结构以来,苯及其衍生物的合成与转化取得了里程碑式的进展。如图1a所示,苯具有六个离域的π电子,因此高度稳定并且所有的碳-碳键键长均相等(1.39 Å)。相比之下,苯的五元环状构造异构体——戊富烯(Pentafulvene,5-亚甲基-1,3-环戊二烯,也称“富烯”)则表现出截然不同的反应性。如果将三共轭碳环的环尺寸进一步缩小,那么就会形成一种不寻常的四元环状构造异构体,即反芳香性的丁富烯(Butafulvene,3,4-二亚甲基-环丁烯)。尽管苯及戊富烯的合成方法较为成熟,但是丁富烯的合成却较为困难,这是因为其固有的高张力能和反芳香性。早在60多年前,Blomquist等人从truxinic酸出发,经八步转化合成了丁富烯(图1b);之后Huntsman等人证明1,5-己二炔可以在350 ℃下异构化为中间体1,2,4,5-己四烯,随后经四电子环加成得到丁富烯,但是反应条件较为苛刻(图1c)。
针对这些问题,浙江大学的麻生明院士、郑剑研究员团队和中国科学院大连化学物理研究所的陈庆安研究员团队设想能否从简单易得的炔丙醇衍生物出发,与炔丙基/联烯基-金属化物进行偶联形成双联烯,再在过渡金属的催化下经环金属化和还原消除得到丁富烯(图1d)。近日,该团队报道了一种高效的钯催化偶联方案,通过炔丙基化合物的反应快速地组装了对称和非对称的丁富烯衍生物,实现了化学版“3”(3碳单位的炔丙基) “3”(3碳单位的炔丙基或联烯基)=“4”(4碳单位的环丁烯) “2”(两个共轭的1碳单位环外双键)。该反应不仅条件温和、底物范围广、官能团耐受性好,而且为高张力环丁烯衍生物的合成提供了一种简单高效的方法。相关论文发表于Nature Chemistry,第一作者为浙江大学化学系博士生黄鑫和中科院大连化物所的陈炳志。
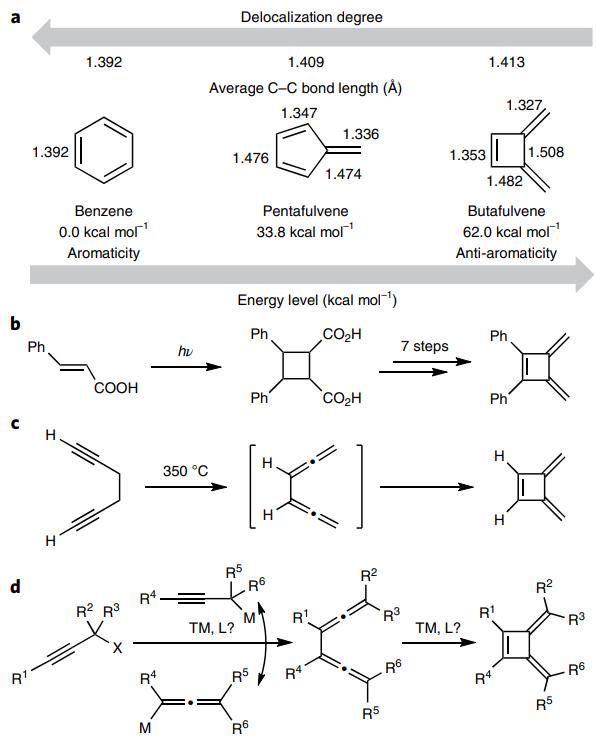
图1. 丁富烯的合成方法。图片来源:Nat. Chem.
首先,作者选择碳酸炔丙酯1a和B2Pin2(1.1 equiv.)为模板底物对反应条件进行筛选,并得到最佳条件:即在Pd(OAc)2(1 mol%)为催化剂、Gorlos-Phos•HBF4(4 mol%)为配体、KHCO3(3.0 equiv.)为碱、H2O(2.0 equiv.,增加碱的溶解度以提高2a/3a的选择性)和THF为溶剂的条件下于40℃反应20 h,能以92%的产率得到目标产物2a,并通过X-射线衍射分析证实了其结构。在最优条件下,作者对炔丙基碳酸酯的底物范围进行了考察(图2,条件A),结果显示带有供/吸电子基团的3-芳基(2a-2f)、3-噻吩基(2g)、3-烷基(2m-2p)甚至3-酯基(2q)取代的炔丙基碳酸酯都能兼容该反应,以中等至较好的产率得到所需产物,尽管底物1q需要更高的催化剂负载量。除了甲基之外,炔丙基取代基还可以是四亚甲基(2h)、五亚甲基(2i、2n)、4-氧杂五亚甲基(2j)、二乙基(2k)和二丙基(2l),其中底物1i能以克级规模进行反应,并以75%的产率得到2i。值得一提的是,当使用3-单取代炔丙基碳酸酯进行反应时,也可以获得相应的产物2r和2s。

图2. 合成对称丁富烯的底物范围。图片来源:Nat. Chem.
然而,一级炔丙基底物却无法在当前的最佳条件下进行反应,作者推测可能是由于一级炔丙基碳酸酯对原位生成的Pd(0)的反应性太低所致。在调整联烯基前体后,原位制备的联烯基-铟试剂可以在Pd(PPh3)4的存在下与3-苯基炔丙基溴4a顺利地进行反应,以91%的产率得到末端丁富烯5a(图2,条件B),并且能以克级规模进行制备(1.03 g,产率:89%)。另外,炔丙基碘化物/氯化物也是合适的偶联配偶体,分别以42%和74%的产率得到5a。随后,作者探索了底物范围,结果显示在苯环对位带有给电子(5b-5e)和吸电子取代基(5f-5h)、间位带有取代基(5i-5k)甚至带有大位阻2,6-二甲基(5l)的底物均能实现这一转化,以42-87%的产率得到所需产物,其中5h的X-射线衍射分析显示环外C=C双键的键长约为1.32 Å,这与乙烯的键长接近(1.33 Å,174 kcal mol-1),而且苯环与丁富烯环不共面,两者之间的二面角分别为22.1°和33.5°。值得注意的是,当使用仲/叔炔丙基溴作为底物进行反应时,也可以形成相应的产物2a和5m,尽管产率较低。此外,带有不同烷基/苯基烷基(5n-5p)以及具有易脱除苄氧基(5r-5w)的底物均能在标准条件下顺利地进行反应。
为了进一步探究反应机理,作者进行了对照实验。具体而言,将底物1a的反应时间缩短至80 min后,没有检测到相关的双联烯中间体。类似地,4a的反应中也没有观察到相关的双联烯中间体,即使在较低温度下进行反应。有趣的是,当1m在最优条件下反应2 h后,能以34%的产率分离出双联烯中间体7m(图3a),同时还在粗反应混合物中观察到联烯基硼酸酯6m和丁富烯 2m。为了阐明7m是否为转化的关键中间体,作者将7m置于最优条件下进行反应,以99%的NMR产率形成2m(图3b)。另外,对照实验表明钯、KHCO3和B2Pin2对2m的形成至关重要。若没有B2Pin2,2m的NMR产率仅为2%,并且2m的产率与B2Pin2的量呈正相关,证实了[B]物种在环化过程中的重要性。基于上述实验结果,作者提出了可能的反应机理(图3c)。首先,Pd(0)催化剂与碳酸炔丙酯1进行氧化加成得到联烯基钯中间体A,再与B2Pin2反应得到联烯基钯中间体B。接着,B经还原消除产生联烯基硼酸酯6并再生Pd(0)。另一方面,另一分子的联烯基钯物种A与联烯基硼酸酯6进行偶联得到双联烯基钯物种C,C经还原消除得到双联烯7。随后,7中的一个联烯单元与LnPd -Bpin(由LnPd和B2Pin2氧化加成、与KHCO3配体交换得到)进行插入得到中间体D,接着经分子内碳钯化得到环丁二烯E,最后释放出LnPd -Bpin便可得到所需产物2。
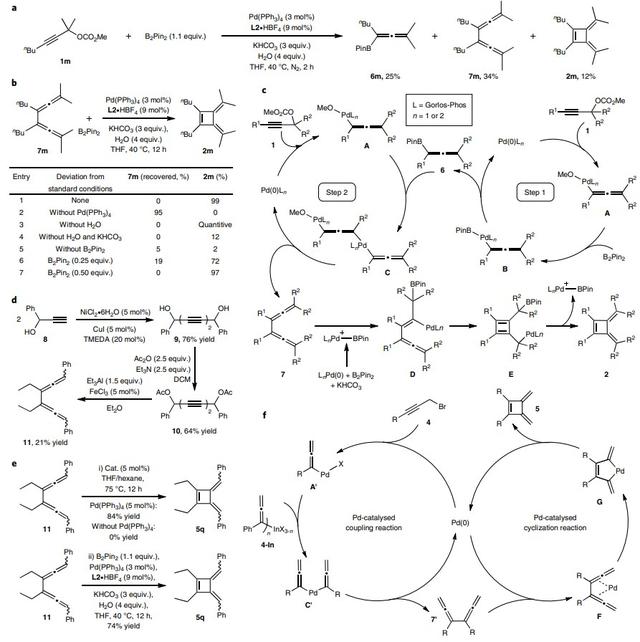
图3. 机理研究及两种策略可能的机理。图片来源:Nat. Chem.
对于涉及铟试剂的方案,作者从炔丙醇8出发,经Glaser-Hay偶联/酯化/双SN2' 偶联三步转化得到双联烯11(图3d),随后将其置于Pd(PPh3)4下于75 °C进行反应,以84%的产率获得所需产物5q(图3e),若无钯催化剂则无法获得5q。另外,将双联烯11置于标准条件下进行反应时,5q的收率可达74%,这些结果表明钯催化剂在环化过程中具有重要作用。基于此,作者提出了可能的反应机理(图3f):首先,Pd(0)物种与炔丙基溴4进行氧化加成得到联烯基Pd(II)中间体A',再和预先形成的有机铟试剂4-In进行转金属化产生双联基-Pd(II)物种C',接着经还原消除得到双联烯中间体7'-Pd配合物F。随后,F经氧化环金属化产生五元钯环物种G,G经还原消除便可得到目标产物5并再生Pd(0)催化剂。
基于上述机理,作者通过碳酸炔丙酯1和联烯基硼酸酯6之间的交叉偶联反应开发了一种获得非对称丁富烯12的途径(图4a)。然而,由于有机铟试剂与炔丙基溴的可逆交换,使用联烯基-铟方案的交叉偶联反应会形成对称和非对称丁富烯混合物并且不可分离。有趣的是,作者开发了5a与芳基碘化物的Heck反应,并以中等的收率和良好的立体选择性获得相应的非对称丁富烯13a-c(E/Z =10:1,图4b)。此外,非对称丁富烯15也可以由叔炔丙基乙酸酯与炔丙基溴化物的反应制备而成(图4c),并且带有不同电性基团的3-芳基(15a-15e)、3-烷基(15g和15h)取代的叔炔丙基乙酸酯甚至仲炔丙基乙酸酯(15k)均能兼容该反应,并以中等至较好的收率得到相应产物。遗憾的是,三苯基取代(15f)或末端炔丙基乙酸酯(15i)却无法实现这一转化。
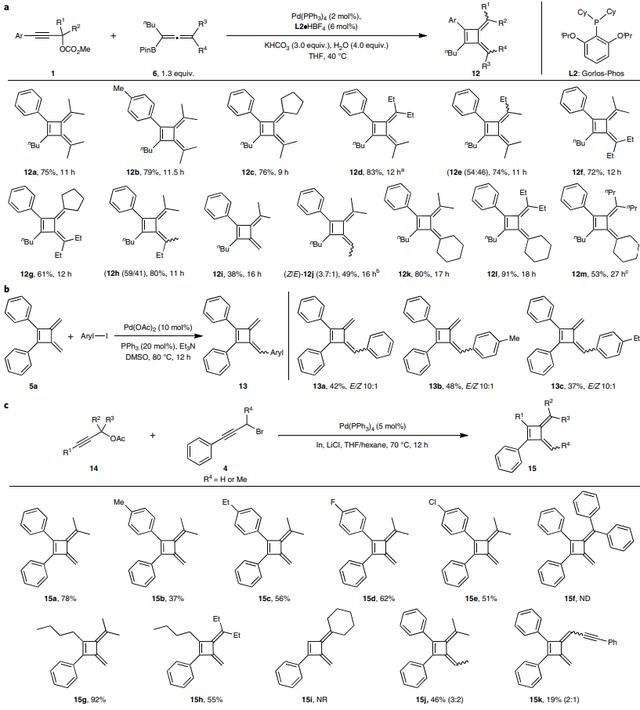
图4. 非对称丁富烯的底物范围。图片来源:Nat. Chem.
最后,作者对产物进行了衍生化。具体而言:1)5a与苯炔中间体发生[2 2]环加成反应得到具有两个四元环的高张力螺环16(图5a),并且其末端C=C键还能进行后续的转化;2)5a与2-重氮-2-苯乙酸甲酯进行环丙烷化反应获得双环产物17(图5b);3)2a中的二烯单元与4-氯/4-溴-苯硫醇之间的可见光诱导反应以良好的产率和专一性得到1,4-加合物18a和18b(图5c),其中18a的结构经X-射线衍射分析证实;4)2a的其中一个exo-C=C键可以在HBr水溶液中进行加氢羟基化,以优异的区域选择性形成相应的产物19a(图5d);5)2o经关环复分解反应以94%的收率得到[4.2.0]-双环产物20,可进一步芳构化为苯并环丁烷21,随后在不同量m-CPBA的存在下发生环氧化反应获得相应的高张力产物22和23(图5e)。
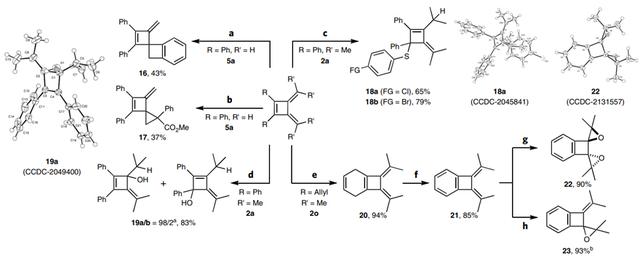
图5. 合成应用。图片来源:Nat. Chem.
总结
本文作者团队使用容易获得的炔丙基碳酸酯或溴化物作为起始原料,开发了一种实用且全面的钯催化高张力对称或非对称反芳香性丁富烯的合成策略。该方法的显著优点包括温和的反应条件、广泛的官能团耐受性和两个简单的催化体系。可以预见,这一策略将促进丁富烯化学的进一步发展。
Palladium-catalysed construction of butafulvenes
Xin Huang, Bing-Zhi Chen, Pengbin Li, Ding-Wei Ji, Jinxian Liu, Hao Zheng, Sa-Na Yang, Yan-Cheng Hu , Boshun Wan, Xiang-Ping Hu, Chunling Fu, Yankai Huang, Jian Zheng, Qing-An Chen , Shengming Ma
Nat. Chem. 2022, DOI: 10.1038/s41557-022-01017-9
导师介绍
麻生明
https://www.x-mol.com/university/faculty/14387
陈庆安
https://www.x-mol.com/groups/qachen
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






