常见的双基配体有哪些(Science报道配体参数化)
本文来自X-MOLNews
2010年的诺贝尔化学奖分别颁发给Richard F. Heck、Ei-ichi Negishi和Akira Suzuki三位科学家,以表彰他们在钯催化交叉偶联反应方面的贡献。钯催化的交叉偶联反应为高效构建多种C(sp2)-C(sp2)键制备复杂的分子提供了一种重要的方法。其中,Suzuki偶联反应(也称“Suzuki-Miyaura反应”)由于底物适用范围广、体系稳定性强、原料易得且毒性低,在医药研发及工业生产中得到日益广泛的应用。

图1. Richard F.Heck、Ei-ichi Negishi和Akira Suzuki获得2010年诺贝尔化学奖。图片来源:Angew. Chem. Int. Ed.
正是由于应用广泛,化学家也很关注Suzuki偶联反应还有哪些需要改进的地方。传统的Suzuki偶联反应能够实现C(sp2)-C(sp2)键的构建,但很难通过烷基硼试剂形成C(sp3)-C(sp2)键,即该方法主要局限于新型平面分子的制备,很难调控分子的立体结构。而从二级烷基硼试剂出发,通过有效的立体控制合成特定立体构型的分子将带来十分重要的应用价值。人们已在二级烷基硼试剂参与的Suzuki偶联反应中做出了大量的努力,反应中加入具有旋光性的烷基硼化合物以得到具有手性的产物。但是该类方法依然存在一些问题:(1)由于C(sp3)-B键高度共价,空间位阻大,转金属化过程比较缓慢;(2)反应倾向于发生β-H消除并重新插入形成烷基钯物种,由此导致烷基异构化并发生立体中心外消旋化。为了克服以上困难,二级烷基硼试剂常需通过引入α位sp2杂化的C、杂原子或具有强配位能力的β-羰基进行活化,然而烷基硼试剂在转金属化时手性中心可能发生立体反转或立体保持,其最终结果会受到底物、催化剂和反应条件的影响。目前人们对转金属化的机理尚不十分明确,如果建立新的模型深刻理解这一过程成为人们思考的问题。
美国纽约市立大学(CUNY)的Mark R. Biscoe教授等人曾利用非活化的二级烷基硼作为亲核试剂,同时以PtBu3作为配体,改进了Pd催化立体定向的交叉偶联反应,得到了立体构型翻转的烷基芳香烃产物。最近,Mark R. Biscoe教授和犹他大学(Utah)Matthew S. Sigman教授等研究者以膦配体为考察参数,通过预测统计模型选择和设计促使反应构型翻转或保持的不同配体,探究Suzuki偶联反应构建C(sp3)-C(sp2)键的转金属化过程,总结了不同膦配体对产物立体构型的影响,并提出该过程的通用模型。他们还利用这一模型实现了立体构型保持的Suzuki偶联反应,并进一步筛选出用于立体构型翻转反应的最优配体,由此从相同的二级烷基硼亲核试剂出发,通过改变膦配体达到产物立体构型调控的目的。相关工作发表在Science 上,共同第一作者为CUNY博士生Shibin Zhao和Utah的Tobias Gensch博士。
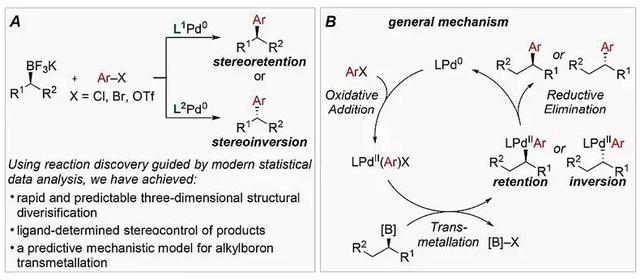
图2. Suzuki偶联反应构建C(sp3)-C(sp2)键及其机理。图片来源:Science
作者首先以吸电子能力不同的取代基修饰的氯代芳香烃与对映体富集的sBuBF3K的Suzuki偶联反应研究底物电子特性对反应结果的影响,发现随着芳香烃电子密度降低,产物的立体翻转程度出现下降,由此说明底物的电子效应对转金属化过程具有一定的影响。当固定反应底物,考察不同膦配体对反应的影响时,作者发现产物的立体构型同样出现显著差异,但其变化与配体的立体效应(立体角)无明显关联。

图3. 不同反应底物和膦配体对产物立体构型的影响。图片来源:Science
为了具体研究膦配体对产物立体构型的影响,作者进一步以对映体富集的sBuBF3K和4-氯苯甲酸乙酯的Suzuki偶联反应为例,考察不同配体对产物立体构型以及支链与线性产物比例的影响进行考察。基于特殊处理具有代表性的构象是描述配体整体性质的重要手段,作者设计了一个流程进行数学建模(图4A):(1)通过分子力学(MM)构象搜索分析膦配体的构象,找到具有代表性的低能量构象;(2)利用密度泛函理论(DFT)对得到的构象进行几何优化;(3)定义四个描述配体的参数研究配体的构象动力学,包括数学上的极限(最大值和最小值)、Boltzmann加权平均数和构象的最低能量值;(4)分析膦配体不同时产物的立体构型分布和支链与线性产物的比例,这两者分别代表转金属化发生产物构型保持与翻转之间的竞争情况和β-H消除/异构化副反应的竞争情况。

图4. 膦配体相关参数的收集与统计模型(A)以及按照该流程优化不同膦配体(B)。图片来源:Science
作者探究了支链与线性产物的比例和最终对映体光学纯度的相关性,发现β-H消除会导致外消旋化和异构化,由此影响转金属化对产物立体构型变化影响的判断。由统计结果可以看出,膦配体的最小宽度(B1)与支链和线性产物的比例具有一定的相关性,证明较大的配体更有利于还原消除反应,从而抑制β-H消除。因此作者仅进一步研究配体对反应的影响。作者以P孤对电子处的最低静电势(Vmin)代表膦配体整体的电子特性,通过简化的数据探究产物对映选择性和配体电子特性的相关性,得出以下结论:(1)体积大的配体能抑制β-H消除和外消旋化;(2)含有吸电子芳基取代基的膦配体有利于反应的构型保持,并加速还原消除过程。基于这些原则,作者研究了不同联芳基膦配体对反应结果的影响,得到了几种效果优异的膦配体,使用强π电子受体配体bis-CF3PhSPhos(11)和 bis-CF3PhXPhos(14)可促进Suzuki偶联反应,并显著提高产物的对映体选择性,产物构型保持,ee值为90%,异构化程度也得到最大程度的抑制,而使用强σ给电子配体9作为配体,产物的立体构型发生最大程度翻转,ee值为-93%。由此说明,反应可通过选择合适的配体来合理预测产物构型的保持或翻转。
接下来,作者使用不同对映体富集的非活化二级烷基三氟硼酸盐与卤代芳香烃设计Suzuki偶联反应,研究该模型在实际应用中的效果。反应以甲苯和水作为混合溶剂,碳酸盐作为碱,不管芳基亲电试剂是富电子、缺电子,还是邻位含有取代基,产物均具有良好的对映选择性。含有噻吩及苯氧基取代基以及位阻较大的烷基硼酸盐也可以顺利参与反应。对映体富集的烷基硼酸盐37可以分别在配体14和9的辅助下得到两种非对映异构体产物17a和17b。

图5. 不同膦配体辅助下的Suzuki偶联反应实现产物的立体构型保持和翻转。图片来源:Science
为了进一步确认膦配体对产物立体构型影响的原因,作者又用数学建模的方式研究了转金属化的机理。以烷基硼酸盐20与氯代芳香烃作为模板底物,考察24种不同膦配体辅助下的反应情况。其中以P-C反键轨道的平均能量Eσ*(P-C)avg代表π反馈键,P孤对电子轨道的能量ELP(P)代表配体提供σ电子的能力,同时引入B1和L两个参数对模型进行修正。从图6B中多元线性回归的结果可知,仅Eσ*(P-C)avg和ELP(P)两个参数就可在一定程度上反映转金属化的过程。结果表明,构型翻转过程主要是因为配体具有强σ给电子能力,由此稳定双配位的阳离子钯中间体;而构型保持则受到π-反馈键的影响。体积较小的配体易导致β-H消除反应,由此引入B1和L两个参数。当将24种膦配体中体积较小的四种舍去,产物的立体构型与Eσ*(P-C)avg和ELP(P)具有良好的线性关系。该多元回归分析方法为膦配体如何影响转金属化的机理提供了良好的解释,也为未来发展交叉偶联反应提供了理论基础。

图6. 利用多元回归建模的方法研究转金属化的机理。图片来源:Science
——总结——
化学是在分子、原子层次上研究物质性质、组成、结构与变化规律的科学,而数学具有化繁为简的能力,在化学中运用数学工具,分析化学变量之间的关系,可以帮助人们从复杂的体系中找到可以遵循的规律,具有事半功倍的效果。该工作将化学问题抽象为数学问题,简化影响的因素,利用数学建模的方式研究了不同膦配体对二级烷基硼酸盐参与的Suzuki偶联反应产物立体构型的影响。基于这一模型,人们可通过选择不同的配体得到立体构型保持或翻转的偶联产物,还可实现在反应前对产物的立体构型进行合理预测。这种方法对今后发展立体选择性的偶联反应具有重要的借鉴意义。
原文
Enantiodivergent Pd-catalyzed C–C bond formation enabled through ligand parameterization
Science, 2018, DOI: 10.1126/science.aat2299
导师介绍
Mark R. Biscoe
http://www.x-mol.com/university/faculty/5761
Matthew S. Sigman
http://www.x-mol.com/university/faculty/1749
(本文由Sunshine供稿)
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






