阐述金属电解精炼的电化学原理(全方位解析钠离子电解液溶剂化结构原理)
第一作者:
Tian Zhengnan(阿卜杜拉国王科技大学(KAUST))
通讯作者:
明军研究员(中国科学院长春应用化学研究所)
Husam N. Alshareef(阿卜杜拉国王科技大学(KAUST))
背景
钠离子电池(SIB)由于钠储量丰富,被认为是后锂时代最有前途的电池技术。在过去的20年里,为SIB探索新的电解质一般都是依靠 "固体电解质间相(SEI)"理论来优化电解质成分。然而,许多观察到的现象不能用SEI理论完全解释。因此,电解质溶剂化结构和电-电解质界面行为最近得到了巨大的研究兴趣,以解释性能的提高。
本工作对SIBs电解液溶剂化结构进行了系统的研究,阐明了具体的溶剂化结构设计准则及其对电化学性能的影响。详细讨论了溶剂化结构形成的关键驱动力,以及调整SIB溶剂化结构的最新进展。相信本综述可以为高性能SIB甚至其他新兴电池系统的电解质优化策略提供新的见解。
虽然,电解质的溶剂化设计非常重要,并引起了广泛的研究兴趣,但目前还没有一篇文章总结电解质工程领域中SIB的溶剂化设计原理和方向。因此,首次从电解质溶剂化行为的角度对SIB行为进行了总结。
一、说明了溶解背后的驱动力(配位键、偶极相互作用、氢键),其中详细分析了电解质基本成分(即金属盐、溶剂、添加剂)的特性及其相互作用(如Na 离子与阴离子的配位、Na 离子与溶剂的配位、溶剂与溶剂的相互作用)。
二、描绘了Na 离子的溶剂化结构,包括几何参数和热力学描述符。更重要的是,解释了溶剂化结构和观察性能之间的相关性。最后,总结了可用于调整溶剂化结构以优化电池性能的策略(图1b)。本综述提出了一个全新的视角,结合理论和实验方法,利用电解质溶剂化设计来改善SIB和其他移动离子电池的性能。

图1
a) 钠离子电池电解质的主要发展和报告成就的年份。b) 显示钠离子电池(SIBs)电解质溶解设计问题的方案。
理想电解质的要求
一、钠盐
金属盐作为电解质的主要成分,在决定SIB的电化学性能方面起着重要作用。金属盐在SIB的电解质中的作用包括以下几个方面。
1)金属盐作为电荷载体的一部分,在两个分离的电极之间进行传输。这些载流子决定了电解质的离子传导性;不良的离子传导性会降低许多电池参数(例如,它可以极大地增加电化学极化)。
2)钠盐会影响SEI的组成,这些组成在循环过程中可能会被溶解和破坏,从而降低SIB的稳定性。
3) 钠盐产生的钠离子参与电极材料内的插层(去插层)反应。因此,钠离子的溶剂化结构,包括几何形态和离域电子密度,可以大大影响钠离子的扩散,特别是通过电极/电解质界面。同样,在散装电解质中,电解质的化学和热稳定性在一定程度上受到钠离子的溶剂化结构的影响。
4) 除了钠离子的导电性、SEI组成和钠离子的溶剂化结构,阴离子氧化时的热力学HOMO能量可以在一定程度上限制电解质的电位窗口,从而限制SIBs的总能量密度。
5)大多数钠盐的化学毒性和腐蚀性对实际应用中的电池安全有重要影响。
鉴于上述考虑,理想的钠盐应具有几个特点:
首先是高溶解度,这可以实现有利的离子传导性。电导率取决于两个参数,包括自由移动的离子的数量和它们的速度。对于前者,离子的总数是由钠盐的溶解度决定的。对于后者,溶剂的特性控制着盐中阳离子和阴离子的移动速度(例如,溶剂的介电常数和粘度)。除了溶剂的介电常数外,值得注意的是,电解质中存在的阳离子和阴离子的价位也在一定程度上影响了移动性。然而,由于Na 离子的单价性质,阳离子/阴离子的价态或氧化状态并不重要。因此,把重点放在盐类的溶解度上。
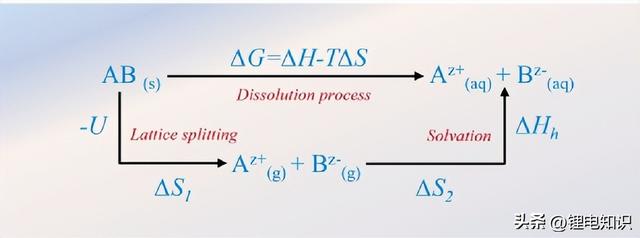
图2溶解过程中所考虑的能量参数。它们包括盐的晶格能(U)和溶解能(ΔHh)。
钠盐的溶解过程可以分为两个单独的过程,包括由晶格能(U)决定的晶格解离(分裂)和由溶解能(ΔHh)决定的与溶剂的溶解。溶解过程的简化Born-Haber循环如图2所示,其中ΔH = -U ΔHh,ΔS = ΔS1 ΔS2。根据这个循环,较小的吉布斯自由能(∆G)代表溶解过程更容易进行。较高的晶格能会降低盐的溶解度,而较高的溶解能会增加其溶解度。例如,在典型的非质子溶剂中,钠盐,如NaCl和NaF,几乎是不溶的。其原因是由于两个电负性差异很大的原子之间的强离子键导致了非常大的U。相反,离子晶体的极化增强(电负性差异减少)将导致离子键转变为共价键,这对盐类在非质子溶剂中的溶解有利。
另一个被广泛接受的概念涉及弱配位阴离子(WCAs),如[CF3SO3]-, [BF4]-, [ClO4]-, [AlX4]-, [MCTFSI]-, 或 [MF6] (X = Cl-I; M = P, As, Sb, etc.) 。在这样的阴离子中,负电荷在阴离子上是离域的,这使我们能够通过利用吸电子基来调节阴离子中的原子的电负性。因此,电荷离域的阴离子和阳离子的相互作用将大大减少,U也将减少,从而提高溶解度。
钠盐的第二个理想特性是电化学稳定性。在理论上,为了保持SIB的热力学稳定性,电解质的电化学窗口(ESW)应该超出阳极和阴极的氧化还原电位。ESW被定义为最低未占分子轨道(LUMO)和最高被占分子轨道(HOMO)之间的差异。有鉴于此,钠盐中阴离子的种类对电解质的ESW有很大影响。例如,在EC/DEC的混合溶剂中,氧化电位遵循NaPF6 > NaClO4 > NaTFSI > NaFTFSI > NaFSI的顺序,这意味着PF6-离子表现出最低的HOMO水平(-11.67 eV),不易失去电子和分解。至于ClO4-离子,HOMO水平位于-7.89 eV,代表氧化过程中的化学稳定性差。然而,化学稳定性是相对的,这表示绝对稳定的阴离子不存在于SIB中。此外,根据以前的报告,钠盐的阴离子可以参与SEI的形成。
例如,PF6-离子在还原过程中的分解产物NaF是SEI的主要成分。简而言之,钠盐中的阴离子可以在两个方面影响电解质的化学稳定性:一个是阴离子的HOMO水平限制了SIB的最高电化学窗口;另一个是阴离子的LUMO水平促进SEI的形成以排除电解质进一步分解。
理想的钠盐的另一个理想特征是具有良好的热稳定性和低毒性。在实际应用中,安全性非常重要。因此,钠盐应具有良好的热稳定性。Eshetu等人研究了钠盐的热稳定性,显示出以下热稳定性趋势。NaClO4 > NaBF4 > NaTFSI > NaPF6 > NaFTFSI > NaFSI。很明显,NaClO4表现出最高的热稳定性。然而,由于NaClO4的强氧化性(给电子的趋势)和在干燥状态下的爆炸性,很少被实际利用。此外,为了广泛地实施SIB,必须考虑到毒性问题。例如,由于有毒的副产品,基于AsF6-和SbF6-的钠盐很少被使用。

图3. 常见钠盐的化学和物理特性。
一般来说,晶格能较小的晶体表现出最高的导电性(NaPF6>NaClO4> NaTFSI>NaOTf>NaBF4)。
HOMO水平趋势:NaOTf > NaClO4 > NaTFSI > NaBF4 > NaPF6,这表明NaOTf和NaClO4更容易被氧化,限制了SIB的电压窗口。
因此,在有机溶剂中溶解的要求筛选了大部分的钠盐。当把氧化性、还原性、热稳定性和毒性结合起来考虑时,可以缩小最合适的钠盐的范围。分析表明,NaPF6提供了最佳的折衷方案。
二、有机溶剂
有机溶剂可以通过以下因素影响电池的性能。
1)作为可溶性钠盐的媒介,电导率受溶剂化学的影响很大。
2)溶剂可能参与功能性SEI的形成,这可能影响库仑效率和循环耐久性。
3)更重要的是,有机溶剂决定了钠离子的溶剂化结构,然后控制电解质和电极之间界面的去溶剂化行为。
4)同样,溶剂分子的电化学稳定性可以限制类似于钠盐的SIB电压窗口。
5)由于有机溶剂是SIB中唯一的液体成分,其不稳定性(挥发性、易燃性)和毒性会影响实际应用。
因此,最佳的有机溶剂应具有以下特点。
高介电常数、低粘度和适度的路易斯酸度/碱度,这可以实现有利的导电性。电导率由自由移动的离子数量和离子迁移率决定。前者是由钠盐的溶解度决定的,它受U和∆Hh的影响。与主要受盐的固有结构影响的U不同,∆Hh主要受溶剂分子和溶质离子之间的相互作用影响。介电常数是一个宏观测量的参数,定义为在电解质中分离离子对的能力,显示出与∆Hh的正相关关系。因此,对于具有相同U的盐来说,具有较高介电常数的溶剂会使盐的溶解度提高,从而实现有利的导电性。溶剂分子的路易斯酸度/碱度(接受/给电子能力)是另一个可以影响∆Hh的参数。
从理论上讲,具有强Lewis碱性的溶剂分子应该促进钠盐的溶解过程。这是因为Na 离子(具有空轨道)和具有孤对电子的溶剂分子之间可能发生配位,从而增加盐的溶解度。然而,如果Na 离子和溶剂分子之间的相互作用太强,可能会导致困难的去溶剂化过程,其中可能导致Na 离子与溶剂分子的共同插层。除了影响溶解度外,溶剂对离子的流动性也起着重要作用。溶剂的粘度体现了分子之间的固有吸引力,对离子迁移率有明显的影响。粘度的降低会使电导率提高一个数量级。
SIB溶剂的另一个理想特性是提高电化学稳定性。与钠盐中的阴离子类似,溶剂分子在充电/放电过程中的氧化和还原与电极的氧化还原反应发生竞争。溶剂的HOMO和LUMO决定了稳定的ESW,也就是说,在电子能量高于LUMO时,溶剂被还原,在电子能量水平低于HOMO时,溶剂被氧化。用溶剂的氧化电位和还原电位之差来表示ESW。要求溶剂的电位差要大。然而,一种更普遍的意见认为,溶剂的分解并不都是有害的。靠近阳极的溶剂的还原可能有利于功能性SEI的形成。被还原的溶剂分子分解成化学活性自由基,形成涂在阳极表面的新化学成分。

图4. SIB中使用的有机溶剂的结构和特性
其他理想的有机电解质特性包括低熔点、高沸点、高闪点、低毒性和低污染影响的高安全性。溶剂的熔点特别重要,因为电池的温度可能是极端的。当周围温度低于熔点时,溶剂的凝结将导致电池失效。例如,EC溶剂的熔点为36.4 °C,由于它在室温下是固体,所以很难单独使用。沸点影响了电解质的挥发性,反过来又影响了SIB的耐用性,因为溶剂的暴晒。此外,由于电池自燃的风险,闪点也很重要。最后,无毒和环境友好的溶剂对于成功的商业化至关重要。
总之,开发最佳溶剂是实现具有良好电化学性能的SIB的一个关键目标。选择最佳溶剂需要全面考虑,包括钠盐溶解、溶剂化/去溶剂化特性、与电极材料的兼容性、实际工作环境,如极低温度或高温和工作电压窗口。
三、添加剂
与LIB类似,电解质添加剂可用于调整SIB的电化学性能。这些添加剂可以通过各种方式影响SIB。
1)对电极/电解质界面的重大影响。根据普遍接受的观点,添加剂参与了影响电化学性能的SEI形成。最近,与SEI效应形成鲜明对比的是,Ming等人提出了另一种创新观点,即添加剂能够改变界面附近阳离子的去溶剂化过程。
2)添加剂可以改变Na 离子的溶剂化结构,从而改变电解质的离子传导性、溶剂和钠盐的电化学稳定性以及粘度。
3)功能添加剂旨在减轻初级电解质的一些特定缺点,如抗过充、抑制可燃性、以及在极低温度下保持工作。
因此,在电解质中引入添加剂应考虑以下因素。
i) 少量。一般来说,添加剂的重量比应保持在5%以下,因为较高的比例会影响原始电解液的组成,这意味着添加剂可能会支配电化学行为。
ii) 添加剂应有利于形成持久的SEI。添加剂在低电位时的分解产物应参与SEI的形成,以减少不可逆的容量和副反应。
iii) 特定功能的添加剂有独特的要求。例如,抗过充电添加剂要求添加剂分子在比正极正常电荷结束电位稍高的电位下可逆地被氧化;阻燃添加剂要求添加剂分子能够终止在气相中负责燃烧反应的自由基连锁反应,以及粘度稀释剂添加剂等。

表1
表1总结了已在SIB中应用的异质电解质添加剂。令人惊讶的是,自从Komaba等人在2011年首次成功研究以来,碳酸氟乙烯(FEC)添加剂是各种功能添加剂中使用最广泛的。这种成功的主要原因是由于FEC的分解在硬碳表面形成稳定的SEI。
与LIB形成鲜明对比的是,碳酸二氟乙烯(DFEC)、亚硫酸乙烯(ES)和碳酸乙烯(VC)添加剂(已经广泛用于LIB)对SIB的性能有不利的影响。然而,Zhang等人发现,VC能有效地抑制SIB中的界面极化,从而在MoO2阳极表面形成更坚固的SEI。除了为促进更稳定的SEI而添加的添加剂,其他添加剂也针对阴极-电解质界面(CEI)。例如,己腈(APN)已被使用,因为与碳酸盐溶剂相比,它具有更强的给电子能力,因此它更容易在阴极材料表面被氧化,形成稳定的CEI。此外,表1中列出了具有阻燃能力、导电性增强、清除剂和过充电的添加剂。值得注意的是,与LIBs添加剂的大量文献相比,关于SIBs中电解质添加剂的报告是零星的。
分子相互作用和溶剂化
一、电解质成分之间的典型相互作用
配位键
短程相互作用被认为是钠离子和溶剂分子中心之间的一种密集力,随着距离的增加而急剧减少。一般来说,配位键作为一种典型的短程力被考虑在内,它描述了一种双价键,其中两个电子都来自同一个原子(见图5a)。通过核和电子对的静电吸引,配位键显示出与共价键相似的高强度。在电解质中,盐类和溶剂之间有可能存在。阳离子,如Na 离子,在失去最外层电子后有空的2s轨道,而一些有机溶剂分子是极端的电子供体,能够捐赠孤独的电子对(例如,己腈)。在配位键电子共享的基础上,溶剂提供孤对电子对的能力决定了配位键的强度。换句话说,有机溶剂的路易斯基决定了配位键的强度,路易斯基越高,相互作用就越紧密。

图5. 描绘电解质中存在的相互作用和结合的示意图。
此外,在基于各种弱配位阴离子(WCA)的钠盐中,阴离子和阳离子之间普遍存在配位键。阴离子上的电子因吸电子基团的存在而离域,导致Na 离子和阴离子之间的配位键强度明显减弱。在这方面,可以通过选择不同的吸电子基团来改变阴离子的供体数量,进而改变电子离域的程度,从而改变配位键的形成能力。
氢键
氢键也是一种短程相互作用,但比配位键弱得多。它描述了一个氢原子与一个电负性较强的原子或基团(X)共价结合,与另一个带有一对孤独电子的电负性原子(Y)之间的静电吸引。如图5b所示,氢键起源于H原子上的偶极取向,由于X原子的强电子吸引能力,从而导致H原子和Y原子之间的静电力。Schroder等人提出了钠盐中的阴离子与溶剂分子之间形成氢键的问题,特别是对于氟化离子或至少有一个孤对的物种(三丁胺、双(草酸)硼酸盐),观察到与溶剂氢原子(如PC中丙烯基的氢原子)的强相互作用。此外,有机溶剂分子和添加剂分子之间可以建立氢键,从而诱导出独特的溶剂化结构,影响SIB的电化学性能。
范德瓦耳斯力
范德瓦尔斯力(有时被称为静电作用)在各种电解质中是相当普遍的,它是由相邻的正负电荷中心之间的静电吸引或排斥产生的。因此,这些键比配位键更弱。典型的范德瓦尔斯力可分为三种不同的相互作用:
i) 偶极-偶极力,产生于两个相邻分子或原子的永久偶极。它大多源于静电吸引和排斥力的平衡,通常在两个极性分子中产生。对于SIB的电解质中使用的非专业溶剂,偶极-偶极力与溶剂的介电常数有关。在0 < ε < 5时,溶剂被认为是非极性的,如DMC、DEC和EMC。当5 < ε < 30时,这些溶剂被认为是中等极性的,如DME、Diglyme和Triglyme。当ε>30时,这些溶剂被认为是极性的,如EC、PC和DMSO。由于强烈的偶极效应,偶极-偶极力在极性溶剂中的范德瓦尔斯相互作用中占主导地位。
ii) 偶极诱导力与偶极-偶极力相似,但其中一个偶极是由近乎永久的偶极诱导的,因为电子云被正电荷中心吸引而变形。这种键的类型在极性分子之间以及极性分子和非极性分子之间被广泛观察到。
iii) 分散力,是所有原子和分子之间的一种普遍的相互作用,源于瞬间的偶极子形成,归因于特定时刻的电子分布不均。
二、溶剂化结构
溶剂化壳
考虑到典型的相互作用,如配位键和范德瓦尔斯力,一个可能的溶剂化结构模型显示在图6中。为了简化模型,一个孤立的阳离子被置于中心位置。在这种情况下,阳离子和阴离子之间的相互作用被忽略了。该模型被画成一个球体,因为我们认为溶剂是一个连续和均匀的介质,包裹着溶质离子。有两个溶剂化壳。第一个溶剂化壳(内圈所示)由于与阳离子的偶极(溶剂分子A)产生的强静电力而形成。一些具有广泛的路易斯碱度的溶剂分子也表现出与阳离子的强坐标作用。由于静电力和配位键之间的协同作用,吸引和排斥达到了平衡,从而形成了第一个溶剂化壳。值得指出的是,一些文献中提到的 "配位溶剂化壳 "是基于与第一溶剂化壳相同的概念。阳离子和溶剂分子之间强有力的相互作用和紧密的联系使阳离子在第一溶剂化壳内移动,而不是自己迁移。

图6. 显示SIB电解质中阳离子溶剂化结构的模型。
与第一溶剂化壳相比,第二溶剂化壳不那么紧凑;它包括部分被抑制的溶剂分子、被吸引的阴离子,甚至包括离子对。同样,自由溶剂分子和自由阴离子也包含在二级溶剂化壳中。值得注意的是,所提出的溶剂化结构是基于离子对可以被溶剂分离的假设。然而,根据Griffiths和Wijayanayake的研究,溶剂的介电常数对离子对和自由离子或分子有显著影响。通常情况下,如果介电常数小于5,预计只有接触离子对会出现。然而,如果容许率大于23,自由离子和溶剂化壳就会出现。
对于容许率在5和23之间的溶剂,离子对和溶剂化壳都存在。此外,除了考虑溶剂的能力外,阳离子和阴离子之间极其紧密的相互作用,如配位键,将急剧增加离子对的数量,从而减少自由离子的数量。简而言之,溶剂化结构是复杂的,包括静电吸引(排斥)、配位、极化和分散过程。在实践中观察到的实际电池行为是所有这些相互作用的总和,其中建立一个模型来帮助我们理解它们的行为和相互作用是必要的。
几何参数
两个关键参数对于描述溶剂分子的几何排列是必要的。一个是配位数,它表示在第一溶剂化壳中围绕阳离子的溶剂分子的数量。第二个是平均键长,它表示中心的钠原子和溶剂的氧原子之间的距离。
Fard等人根据M06-2X/6-311 G(d,p)水平的优化理论,提出了Na 离子在不同溶剂中的溶剂化几何形状(如图7所示)。例如,在EC溶剂中,每个Na 离子周围有五个溶剂分子;相比之下,由于VC分子的尺寸较大,每个Na 离子周围有三个VC溶剂分子。此外,由于溶剂分子之间的立体阻碍减少,Na -O的平均键长从2.359 Å(EC)减少到2.195 Å(PC)。立体阻碍可以理解为第一溶剂化壳中相邻溶剂分子之间由于偶极子指向同一方向而产生的排斥力。如图6所示,立体阻碍(F,红色箭头)可以分为两个力,一个是沿圆的切线方向(F2,蓝色箭头),另一个是沿离心方向(F1,紫色和橙色箭头)。很明显,F2被相邻的溶剂抵消了。然而,F1力是叠加的(它们加起来),这使溶剂远离阳离子。
因此,大的溶剂尺寸或第一溶剂化壳中的高配位数将导致大的立体阻碍,从而增加Na 离子和溶剂之间的键长。此外,模拟结果表明,随着EC加入到PC中,两个PC和三个EC分子将留在第一溶剂化壳中。对于EC/DMC混合物,两个DMC和四个EC分子占据了第一溶剂化壳。同样,EC/EMC和EC/DEC溶剂都表现出类似的行为,这表明与DEC、DMC和EMC(通常具有相当弱的偶极)相比,EC与Na 离子表现出更紧凑的相互作用。加入EC后,平均键长(溶剂的中心钠原子和氧原子之间的距离)也会发生变化。由于立体阻碍的增加,EC:PC溶剂混合物中的键长从2.233 Å增加到2.321 Å。相反,EC/DEC、EC/DMC和EC/EMC显示出键长的减少,这是由于大尺寸的DEC、DMC和EMC被小尺寸的EC分子取代而产生的立体阻碍的减少。

图7. 不同溶剂中Na 离子的第一溶剂化壳的几何形状
除了溶剂分子的影响外,居中的阳离子本身也会对几何溶剂化结构产生深刻的影响。Pham等人分析了EC溶剂中不同阳离子(Li 离子、Na 离子和K 离子)的第一溶剂化壳。模拟结果显示,Li 离子表现出一个明确的第一溶剂化壳,而较大的Na 离子和K 离子则表现出更无序和灵活的溶剂化结构。这些结果说明,配位数和平均键长都随着阳离子大小的增加而增加。阳离子和溶剂分子之间的相互作用主要是静电的。因此,阳离子的半径越大,阳离子和溶剂的氧原子之间的作用力就越弱,这反过来又诱发了无序和灵活的结构(包括配位数和键长的增加)。
描述SIB电解质中溶解行为的另一种方法是使用热力学变量。这些变量包括
1)结合能(ΔEb),它反映了阳离子和溶剂分子之间相互作用的强度。它可以根据溶剂化复合物和构成复合物的成分(溶剂分子(SM)和Na 离子)之间的能量差异来计算。

2)溶解自由能(ΔGsol),指在某一热力学过程中,减少的内能可转化为外功的部分。ΔGsol<0时,溶剂化过程更容易发生。
对于溶剂化过程,ΔGsol可以用以下公式计算。

3)LUMO和HOMO能级(LUMO/HOMO),表示溶解的Na 离子溶剂的最高占有分子轨道(HOMO)和最低未占有分子轨道(LUMO)。HOMO和LOMO之间的能量差被定义为带隙。
表2总结了钠离子在不同碳酸盐溶剂中的热力学描述符,这是基于Fard等人所做的模拟。显然,在所有单组分溶剂中,Na 离子和EC分子之间的结合能最大(-115.73 kcal mol-1),表示溶解过程中的强相互作用。相比之下,DEC表现出最小的结合能,为-77.02 kcal mol-1,表明溶解过程很弱。仿真结果与以前的分析相一致,即溶解作用显示出对溶剂分子偶极矩的明确依赖性。此外,ΔGol也说明了一个类似的结果,在EC溶剂中Na 离子的溶解过程更容易发生(-71.63 kcal mol-1)。Fard等人进一步澄清说,在大多数溶剂中,ΔGsol与介电常数成正比。这意味着提高溶剂的介电常数将有利于Na 离子的溶解过程。有趣的是,由于VC分子的介电常数非常大,因此不符合这种关联性。

此外,在轨道能和状态密度(DOS)计算的基础上,表2中给出了几种溶剂的HOMO和LUMO能级(溶剂化后)。值得注意的是,所有的Na -溶剂复合物在形成溶剂化结构后都表现出一个负移位(即与孤立或裸溶剂相比,HOMO较低)。这种转变表明,溶剂分子在与Na 离子溶合后变得更耐氧化。另外,我们看到Na -DEC复合物显示出良好的热力学稳定性,有13.85eV的大带隙,而Na -VC显示出较差的热力学稳定性,有8.87eV的小带隙。
为了研究溶解过程,Okoshi等人利用不同的模拟方法(B3LYP/cc-pVDZ(-PP))来计算Na 离子在各种有机溶剂中的溶解能(∆Esol)。事实上,如前所述,溶解能的大小与结合能相等但符号相反。结果表明,Na 离子的∆Esol是:APN(186.3 kJ mol-1)>DMSO(169.1 kJ mol-1)>PC(157.3 kJ mol-1)>EC(151.9 kJ mol-1)>DEC(147.5 kJ mol-1)>ATN(乙腈,137.4 kJ mol-1) > NM(硝基甲烷,118.1 kJ mol-1)。显然,普通碳酸盐表现出适度的溶解能,被认为是溶解和去溶剂化过程的有利溶剂候选。通过拟合和分析,Okoshi等人进一步得出结论,化学硬度(η)、静电势(σ)和∆Esol之间存在线性关系。

这些关系表明,增加静电势或降低溶剂分子的化学硬度,为改善溶解能提供了可行的途径。
溶剂化过程对电化学性能的影响
溶剂化过程可以分为三个独立的步骤(图8)。
1. 其中包括钠离子在有机溶剂中的溶解和溶剂化过程(步骤I);这一步对电解质的电导率有明显的影响。
2. 接下来会发生钠离子在有机溶剂中的迁移过程(步骤II);这一步对于确定电解质电导率也是至关重要的。
3. 是钠离子在电解质和电极之间的界面上的去溶剂化过程(步骤III);这个过程极大地影响了离子的插层行为。
值得指出的是,这三个步骤都是由阳离子、溶剂分子和阴离子之间的相互作用驱动的。例如,在第一步中,盐中阴离子和阳离子的相互作用决定了盐的晶格能,阳离子和溶剂分子的相互作用决定了溶剂化能。在这方面,一方面,开发了周旋于阴离子的钠盐,以提高溶解度。另一方面,研究了具有高供体数的溶剂来提高盐的溶解度。

图8. 溶剂化过程对电化学性能的影响。
在步骤二中,钠离子的流动性也受制于阳离子和阴离子以及溶剂和溶剂之间的相互作用。阳离子和阴离子之间的强烈配位会抑制溶解的钠离子的运输。同样,溶剂分子之间强烈的相互作用,如氢键,将导致粘度升高,反过来抑制离子的流动性。然而,步骤I和步骤II都只影响电解质的导电性,从而导致SIBs的速率性能下降。相反,步骤III是一个更复杂的过程,它包括去溶剂化过程,溶剂和阴离子在表面的分解,跨过能量屏障和通过SEI。这些过程成为决定速率的步骤,极大地影响了容量、速率性能、电压窗口、库仑效率和循环稳定性。
调节溶剂化结构以优化电池性能
电解质的溶剂化结构基本上由阳离子、溶剂分子和阴离子组成。因此,溶剂化壳可以通过调整这三种成分来改变不同成分之间的相互作用,进而对电化学过程的第一、第二和第三步骤产生影响。

图9. SEI理论和溶剂化理论。
总之,根据传统的SEI理论,优化电解质的主流策略,如添加添加剂、使用多种溶剂和改变阴离子,已经被认为有助于形成更好的SEI。然而,电解质中溶剂化结构的影响也不应被忽视。添加剂与阳离子、阳离子与溶剂、添加剂与阴离子、阴离子与阳离子之间的内部相互作用可以改变溶剂化结构,这对电极性能有很大的影响,例如影响石墨性能的Li -溶剂相互作用的强度,即可逆的Li (去)插层与Li -溶剂共同插入导致石墨剥落。
调整阴离子
优化溶剂化结构的最有效方法之一是调整电解质中的阴离子。阴离子和阳离子之间强烈的静电作用会导致离子对的形成,从而抑制阳离子的流动性。同时,离子对可以促进离子团的形成,导致更紧密的溶剂化壳,抑制阳离子的去溶剂化过程。因此,调节阴离子以打破离子对之间的强相互作用,有助于改善电化学性能。
阴离子对溶剂化结构的影响

图10. 使用不同阴离子基盐的电解质的电化学性能。
阴离子在溶解过程中起着关键作用,SEI形成理论虽然重要,但可能不是唯一可以用来调整电极稳定性的因素。因此,通过调节阴离子的行为来优化电池的性能有很大的空间。
弱配位阴离子
如第2节所述,WCAs表现出阴离子上负电荷离域的重要特征。这一特点使阳离子和阴离子之间的配位键变得更弱。一方面,较弱的配位键导致盐类的溶解度提高,晶格能降低。另一方面,从溶解的角度来看,较弱的配位会减少离子对,有利于电解质中松散的溶剂化壳。
WCA的设计原则是相对严格的。
首先,WCA的电荷应该是低的,最好是单价的,以削弱与阳离子的静电作用。在这方面,大尺寸的阴离子将最大限度地减少残余的库仑吸引力,有利于在低极性溶剂中的溶解。
第二,电荷必须在整个实体上高度分散,不应有碱性(热力学)或亲核性(动力学)的位点,因为它们是典型的配位点,可能代表了走向离子配对和进一步WCA降解的第一步。
第三,WCA只能由化学稳定性强的部分构成,以承受与非常活泼的阳离子/中间体的合作。
最后,WCA表面的极化率应该很低。这些基本要求常常导致使用氟化实体作为WCA的构建单元,因为它们通常满足所有的要求。

图12. 为SIBs设计新的WCAs。
WCAs是提高动力学性能的最有效的策略之一,不仅在体电解质中,而且在界面上。此外,除了有益的配位特性外,一些研究表明,大多数WCAs表现出卓越的氧化稳定性,这使得这些盐类能够与高压电极材料兼容。这是因为阴离子在溶剂化结构上的差异可以改变电极表面的界面模型(即阴离子、溶剂、阳离子和电极之间的相互作用)。然而,到目前为止,由于复杂的合成过程和多种设计要求,新WCAs的开发需要的进展非常缓慢。因此,需要研究为非水电解质设计新的WCAs,因为尽管有挑战,但它有很大的潜力。
阴离子受体添加剂
除了调整阴离子的配位结构和电子离域程度外,引入阴离子受体添加剂是调节溶剂化结构和提高电化学性能的另一个途径。一般来说,阴离子受体是一类有机配体,可以通过π-π配位键或氢键在电解质中与阴离子和负电荷官能团(如羧酸盐和磷酸盐)有效而有选择地配位。受体添加剂与阴离子之间的强相互作用,削弱了阴离子与阳离子之间的静电作用,从而减少离子对的形成,提高离子迁移率。然而,一个关键的缺点是,由于添加了阴离子受体添加剂,电解质的分解作用增强;这种分解作用使SEI层变厚,反过来破坏了金属离子电池的速率性能。考虑到这一点,需要改进配方,使阴离子受体添加剂的功能最大化,并将其对速率性能的负面影响降至最低。

图13. 关于在LIB中使用阴离子受体的部分例子。
简而言之,阴离子和阳离子之间的相互作用可以通过添加阴离子受体添加剂来调控。溶剂化结构的改变明显地提高了阳离子的流动性,反过来又改善了电化学性能。此外,阴离子受体中的氟化基团有利于有利的SEI,以抑制金属枝晶。然而,在阴离子受体添加剂上仍然存在一些潜在的问题,如加速电解质的分解。此外,源于阴离子受体添加剂需求的优化溶剂化结构的详细机制还没有被完全理解,特别是从界面排列和去溶剂化过程的角度。
调整溶剂分子
由于溶剂分子在第一溶剂化壳中的主导地位,调节溶剂分子似乎是优化溶剂化结构的一个更直接的策略。溶剂分子针对阳离子的配位能力基本上决定了去溶剂化过程。一般来说,强的配位作用(或偶极)不可避免地导致阳离子-溶剂共插层,如石墨阳极中的Li -溶剂(通常描述为电解质不相容)。阳离子-溶剂共插是非常不可取的,因为它导致电极材料的结构破坏(即石墨剥落),导致电化学性能下降。此外,固相中的插层溶剂分子在充/放电过程中被分解,释放出气体并加剧副反应,反过来又大大损害了电池的性能。然而,由于离子对形成的概率增加,阳离子和溶剂之间非常微弱的相互作用也对电池性能有害,从而排除了阳离子在大量电解质中的迁移。
溶剂对溶剂化结构的影响

图14. 不同溶剂型盐类在SIB中的溶解行为。
多溶剂电解液
早期优化溶剂成分的工作主要涉及调整溶剂类型,通常是将各种溶剂组合成二元和三元溶剂。根据Bommier等人的研究,最常用的钠离子电池电解液是EC/DEC。由于这两种溶剂的优势互补(EC的高介电常数和DEC的低粘度),这些溶剂混合物可以实现良好的电化学性能。不幸的是,很少有报告解释SIBs在多种溶剂存在下的溶解过程。最近,Cao等人提出了一种溶解机制来解释在锌离子水电池中使用二元DMSO/H2O溶剂的优势。如图14e所示,由于DMSO的Gutmann供体数(29.8)高于H2O(18),所以DMSO溶剂部分取代了第一溶剂化壳中的H2O分子。DMSO与Zn2 的优先溶解和H2O-DMSO的强相互作用抑制了H2O的分解。此外,溶解DMSO的分解有利于良好的SEI的形成。这些结果表明,利用多种溶剂可以成为优化溶剂化结构的有效策略,进而实现有利的电化学性能。
调整溶剂的数量
除了使用多种溶剂外,另一个广泛使用的策略是通过调整溶剂分子的数量来优化溶解行为和电池性能。溶剂分子的数量可以通过调整浓度来改变。如图15a所示,传统的电解质(通常使用1M的浓度)保持了阴离子和溶剂数量的平衡,这可以实现有利的离子导电性和适度的电压。然而,由于在电极和电解质界面附近的游离阴离子和溶剂的分解,在电压升高时可能发生副反应。随着电解质浓度的增加(图15b),Na 离子的溶剂化结构发生变化。由于溶剂分子的数量不足,阳离子和阴离子倾向于形成离子对,阴离子发生聚集。由于界面附近的溶剂分解减少,自由溶剂分子数量的减少扩展了电位窗口。此外,如果考虑Na 离子的去溶剂化过程,高浓度的电解质改善了Na 的扩散过程,因为几乎没有溶剂的相互作用。同样,与传统的电解质不同,SEI主要由阴离子的分解产物组成。尽管高浓度电解质显示了一个扩展的电压窗口和快速的去溶剂化过程,但其高成本和有限的离子传导性(高粘度)是主要的限制。因此,有人提出了一个相反的策略,即使用超低浓度电解质。

图15. 不同电解质浓度下的溶解和界面模型

图16. 超高和超低浓度电解质的图示。
溶剂化结构的表征方法
由于溶剂化结构的抽象性,直接可视化的表征技术还不成熟。然而,随着光谱技术以及理论模拟的发展,解读溶剂化结构变得越来越可行,如傅立叶变换红外光谱(FTIR)、拉曼光谱和核磁共振(NMR)。

图18. 拉曼和FTIR对溶剂化结构的表征。

图19. 溶剂化结构的核磁共振表征。
理论模拟
尽管光谱表征可以给出溶剂化结构内部的定性相互作用,但它只反映了大块电解质中相互作用的强度。然而,有时需要考虑电解质成分的局部配位环境和界面情况。因此,理论模拟提供了一个有效的途径来分析溶剂化结构。通常,最常用的是溶剂化结构领域的径向分布函数(RDF)。用g(r)表示,它定义了在与另一个被标记的粒子相距r的距离上发现一个粒子的概率。凭借RDF,可以确定阳离子的溶剂化结构,包括配位数、阴离子的占有率和添加分子的占有率。从RDF衍生出来的潜在平均力(PMF)是另一种有效的方式来表达获得溶剂分子或阴离子进入溶剂化层的能力。配位强度和能量屏障可以通过PMF曲线中的接触最小值得到。此外,埋藏体积(Buried Volume)可以反映溶剂化结构中的立体阻碍和相互作用。除了分子动力学的模拟,溶解离子的电子结构可通过投影部分状态密度(PDOS)进行研,从中得到能级和轨道的信息,进而解读成键信息。此外,热力学参数,如去溶剂化能、结合能、HOMO/LUMO等,也被用来确认溶剂化行为。
Electrolyte Solvation Structure Design for Sodium Ion BatteriesAdvanced Science ( IF 16.806 ) Pub Date : 2022-06-05 , DOI: 10.1002/advs.202201207Zhengnan Tian, Yeguo Zou, Gang Liu, Yizhou Wang, Jian Yin, Jun Ming, Husam N. Alshareef
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






