FQCDSI 项目计划(系列文章基于QbD的2.3.S.2.6申报案例)
前言
在第四篇,通过多变量DoE,建立步骤1(步骤1-反应,步骤1-结晶)的设计空间。本篇继续阐述步骤2(步骤2-反应,步骤2-结晶)设计空间的建立,进而输出整体设计空间和控制策略。
5)-1-2 步骤2
5)-1-2-1 步骤2-反应
在步骤2中,CP-7与CP-8在二氯甲烷中反应,得到Sakuramil粗品。反应后的混合物经淬灭、萃取,并用乙醇进行溶剂蒸馏交换,最终从乙醇和水的混合溶剂中结晶得到Sakuramil成品。
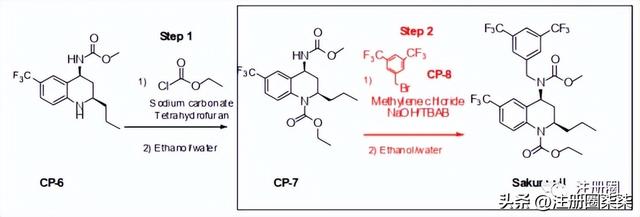
图-2.3.S.2.6-20步骤2工艺
结合对反应的开发,去向和清除数据表明粗品中CP-8的水平为1.2%,存在原料药质量不符合0.10%要求的风险。因此,将CP-8作为限制性试剂(反应中控制当量的试剂)并严格监控反应终点。基于存在多个失败边缘的开发知识,将其评估为高风险,成为设计空间的一个限制性因素。
CP-8的质量属性:选择了一批杂质水平很高的CP-8进行多变量实验,以凸显设计空间控制该起始材料中杂质的能力。这些数据是对实际去向和清除数据的补充,以帮助评估风险并确定可接受的水平和关键性。
反应:
CP-8被用来作为限制性试剂,使用PAT监控CP-8的终点。此外,在探究实验设计中的范围时,对其他总杂质进行监测,以确定(1)是否有新杂质产生,(2)工艺已知微量杂质水平是否增加(<0.1%)。
通过风险评估,对潜在影响原料药中CP-8(和其他总杂质)含量的参数进行了识别。CP-8的化学计量、分相比和反应浓度是对原料药潜在质量属性影响最高。设计了一个实验策略,以确定该工艺参数对这些质量属性的控制,提高工艺的理解和稳健性,并为步骤2-反应的制造工艺建立设计空间。相转移反应的历史数据/知识表明,在开发范围内,NaOH浓度和TBAB催化剂对质量或速率没有影响。
规模和设备考虑(在进行多变量DoE以建立设计空间之前)
与许多相转移反应一样,如果水相和有机相没有得到充分分散,反应速率将受到严重影响,然而,即使在混合不良条件下反应24小时,产品纯度似乎也不受影响。这表明该反应足够稳健,能够承受规模上可能遇到的任何混合问题(例如搅拌暂停)。

显示了使用1000转/分钟(红色)、500转/分钟(蓝色)和200转/分钟(绿色)搅拌速率的实验中CP-7(wt%)HPLC残留结果。
图-2.3.S.2.6-21 搅拌速率与残留CP-7之间的关系(CP-703458)
正如预期的那样,混合对相转移反应速率影响较大,在较高的搅拌速率下,相转移反应进行得更快。如果不能实现水相和有机相的充分分散,就像实验中200转/分的情况一样,即使在24小时后,反应也可能无法进行到底,但在提高搅拌速率后,1.5小时内即可反应完全。
尽管出现这些结果,该反应似乎足够稳健,在所有的实验中,所有进行到底的反应都有典型特征杂质谱,预期在规模放大时不会出现与混合相关的纯度问题。
进行了额外的影响因素实验,以理解实验室和商业化生产时间的差异是否对质量属性有任何影响。这将在反应和结晶的设计空间中得到确认。此外,多变量实验模拟了商业设备及其局限性的 “最差条件”。
Doe探究参数和范围
设计并实施一个三参数响应面设计:
表-2.3.S.2.6-10 步骤2-反应DoE
|
参数 |
低 |
中 |
高 |
|
CP-8当量(相对于CP-7) |
0.9 |
1.05 |
1 |
|
分相比(NaOH/DCM) |
0.25 |
1 |
1.25 |
|
反应浓度(L/kg,DCM体积/CP-7) |
0.25 |
3 |
5 |
反应结论
- 在所有的实验中,CP-8的最高含量均低于1%(1.2%是CP-8的标准,在结晶步骤中可纯化至0.10%)。
- 去向和清除数据表明,5%的未反应CP-7被充分清除至低于0.1%的非特定杂质。此外,选择设计空间中高水平分相比(水相占比高),对5%的CP-7进行清除,也符合0.10%的非特定杂质水平。
- 在参数多变量实验和/或影响因素实验中,没有观察到验收标准的偏离,也没有观察到失败边缘(EoF)。三个参数均处于高水平时,CP-8残留为1%。CP-8的接受标准是1.2%。参数均处于高水平是不切实际的,故该参数为非关键。

图-2.3.S.2.6-22 残留的CP-8和反应工艺参数之间的关系
5)-1-2-2 步骤2-结晶
通过风险评估,对潜在影响原料药中CP-8(和其他总杂质)含量的参数进行了识别,见下表2.3.S.2.6-11。设计了一个实验策略,以确定该工艺参数对这些质量属性的控制,提高工艺的理解和稳健性,并为步骤2-结晶的制造工艺建立设计空间。
规模和设备的考虑
众所周知,结晶与规模和设备相关,然而,这个产品的物理特性被确定为非关键性。结晶的设计将集中在潜在杂质CQA上。执行影响因素实验,以理解实验室和商业化生产时间的差异。收集整个多变量实验中4种遗传毒性杂质的响应数据,确认提出的控制策略。
Doe探究参数和范围
27-3部分因子设计用于结晶研究。在步骤2-反应的DoE中,CP-8的最高含量为1.2%。在结晶研究中加入3%的CP-8(步骤2-反应的DoE中最高含量的2或3倍),冷却速率和去离子水浓度被识别为控制CP-8的关键工艺参数(CPP)。
表-2.3.S.2.6-11 步骤2-结晶DoE
|
参数 |
低 |
标准 |
高 |
|
冷却速率(℃/min) |
0.15 |
0.36 |
0.5 |
|
最终温度 |
14 |
18 |
24 |
|
最终浓度(L/kg,乙醇体积/CP-9) |
3 |
4.5 |
8 |
|
滴加时间(min) |
15 |
30 |
60 |
|
去离子水浓度(%w/w,水体积/乙醇) |
20 |
28-32 |
35 |
|
搅拌速率(rpm) |
150 |
250 |
350 |
|
滴加水前的保持时间(hr) |
2 |
3 |
影响因素参数 |
结晶DoE结论
- 如图2.3.S.2.6-23所示,杂质含量随着去离子水浓度和冷却速率水平的增高而增加。因此,高水平去离子水浓度和高水平冷却速度组合被确定为CPP。公式如下:
- 35%去离子浓度 0.5℃/min冷却速率=杂质CP-8>0.10%的风险
- 在所有的实验中,没有观察到任何来自步骤2-反应的杂质CP-8和CP-7含量超过0.1%。
- 值得注意的是,在提出的设计空间内没有观察到新的杂质。

图-2.3.S.2.6-23 残留的CP-8和结晶工艺参数之间的关系
规模和设备
在分离前,通过改变冷却率和延长接触时间的影响因素实验,确证了杂质CP-8的潜在失效。因此,设计空间包含对任何新设备的温度控制评估,并证明它可以控制冷却速率,从而符合CP-8的质量标准。
CP-6潜在遗传毒性杂质(PGI)数据
表2.3.S.2.6-12显示了在步骤1和步骤2的多变量设计中检测到的PGI的最高含量数据。
表-2.3.S.2.6-12 提出的控制策略的PGI数据支持
|
In CP-6 (SM) |
In CP-7 (步骤1) |
In Sakuramil (步骤2) | |
|
CP-6 |
N/A(98%) |
<200ppm |
<10ppm |
|
CP-3 |
0.1% |
<10ppm |
<1ppm |
|
CP-4 |
0.3% |
<10ppm |
<1ppm |
|
CP-5 |
0.1% |
<10ppm |
<1ppm |
- CP-4, CP-5, CP-3的控制策略:
在CP-6中:高风险的物料属性CP-4(≤0.3%) CP-5和CP-3(各≤0.1%),在Sakuramil中,这三种物质的总量为≤10 ppm。
- CP-6(起始物料)的控制策略:
当通过步骤1和2的设计空间进行制造时,Sakuramil中的含量小于10 ppm(CP-6作为Sakuramil原料药的关键质量属性:≤10 ppm)。
因此,整体潜在遗传毒性控制策略=综合上述两个控制点确保在原料药Sakuramil中的CP-5,CP-3,CP4和CP-6的总和<25 ppm。
5)-1-2-3 步骤2反应和结晶初始关键性风险评估
表-2.3.S.2.6-13对步骤2反应和结晶的多变量分析的输出进行了总结。
表-2.3.S.2.6-13 步骤2多变量分析结果总结
|
参数 |
设计空间 |
标准条件 |
参数或属性关键性论证 |
|
CP-8当量(相对于CP-7) |
0.9-2摩尔 |
1.05 |
非关键* |
|
分相比(NaOH/DCM) |
0.25-1.25 |
1 |
非关键* |
|
反应浓度(L/kg,DCM体积/CP-7) |
0.25-5 |
3 |
非关键* |
|
冷却速率(℃/min) |
0.15-0.5 |
0.36 |
关键。理由:高水平冷却速率与高水平的去离子水浓度组合。 |
|
最终温度 |
14-24 |
18 |
非关键:对CP-8残留无影响。 |
|
最终浓度(L/kg,乙醇体积/CP-9) |
3-8 |
4.5 |
非关键:对CP-8残留无影响。 |
|
滴加时间(min) |
15-60 |
30 |
非关键:对CP-8残留无影响。 |
|
去离子水浓度(%w/w,水体积/乙醇) |
20-35 |
28-32 |
关键。理由:高水平冷却速率与高水平的去离子水浓度组合。 |
|
搅拌速率(rpm) |
150-350 |
影响因素参数 |
非关键:对CP-8残留无影响。 |
|
滴加水前的保持时间(hr) |
2h以上 |
影响因素参数 |
非关键:过滤前延长保持时间,杂质含量没有变大。 |
*在参数多变量实验和/或影响因素实验中,没有观察到验收标准的偏离,也没有观察到失败边缘(EoF)。三个参数均处于高水平时,CP-8残留为1%。CP-8的接受标准是1.2%。参数均处于高水平是不切实际的,故该参数为非关键。
5)-1-2-4 步骤2多变量总结
图-2.3.S.2.6-24对步骤2的多变量分析结果进行了总结:
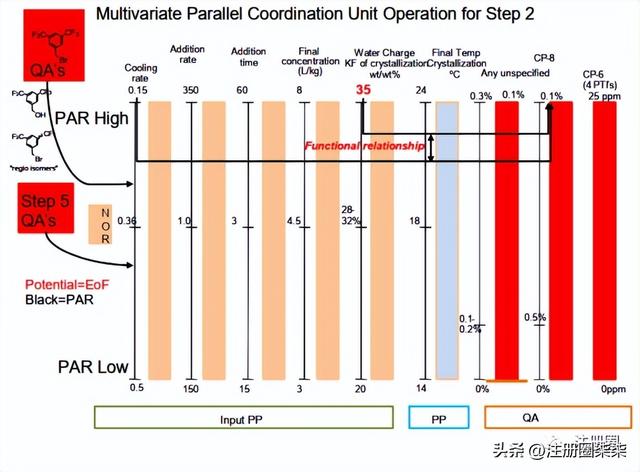
图-2.3.S.2.6-24 步骤2的单元操作变量组合
6)制造工艺关键性评估:最终设计空间和控制策略总结
以下是最终风险评估结果,总结了整体制造工艺设计空间和对每个被识别的关键工艺参数以及关键质量属性的控制策略。
- 高风险(和中风险)工艺参数范围将在S.2.2的制造工艺描述中作为承诺提出。
- 与原料药CQA功能相关的起始物料或原料的物料属性将在S.2.3中定义,并附有可接受标准。
- 中控或分离中间体的重要物料属性将在S.2.4的中间体质量标准中定义,并附有可接受标准。
表-2.3.S.2.6-14 整体制造工艺的控制策略和设计空间总结
|
物料属性/原料药CQA |
控制策略 |
设计空间 |
|
在原料药中,乙基同系物≤1.0% |
|
步骤1的设计空间显示,乙基同系物的可能的最高含量(即使在影响因素条件下)为0.3%。在步骤1的设计空间中没有发现失败的边缘。这是一个非常稳健的工艺,步骤1的反应没有识别出CPP。 |
|
其他总杂质不超过5%(步骤1,中间体物料属性)和非特定杂质0.10%(步骤2,原料药CQA) 中间体物料和原料药 CQA的功能相关。 |
|
众所周知,水(反溶剂)会增加杂质的含量。该参数的NOR在步骤1&2中都是28-32%; · 步骤1和步骤2中的用于结晶的去离子水浓度在高水平是CPP。 (1)步骤1中CCP为50%浓度 (2)步骤1中CCP为35%浓度 |
|
在原料药中,CP-8≤0.10% |
在PAT失败的情况下,作为中控分析,HPLC也可用于确定CP-8的1.2%残留水平,原料药中CP-8的HPLC含量标准订为≤0.10%。
如果反应完成时,CP-8≤1.2%,结晶设计空间显示原料药中CP-8<0.10%。 |
CP-8:结合步骤2的结晶设计空间,用中控PAT方法,证实了RTRt可以将这种杂质控制在<0.10%。 步骤2被确定为一个关键步骤。两个CPP被确定:冷却速率和去离子水浓度。 |
|
遗传毒性杂质(GTI):原料药中共有4种GTI,总量不超过25 ppm |
(1)CP-4(≤0.3%) (2)CP-5和CP-3(各0.1%) |
数据表明,如果这3个杂质符合CP-6中每一项的质量标准,CP-6在原料药中的含量小于10ppm,那么4种GTI的总量不会超过25ppm。 合理性。这些杂质具有极高的亲脂性,并且具有强大的清除因子。去向和清除实验表明,杂质即使在CP-6中的含量为1%,总量仍然远远低于TTC。 |
|
手性(对映体和非对映体) 在原料药中,不超过 0.10% |
(1)≤1%的对映体 (2)≤1%的非对映体
应注意,原料药的分析方法对立体异构体是专属的,手性杂质作为"非特定杂质"控制。 |
通过设计空间内的去向与清除,所有的对映体和非对映体含量远低于原料药中0.10%的标准。 |
规模与设备
1. 步骤1不依赖于规模和/或设备。规模和设备的变更将通过质量体系进行管理。
2. 步骤2中仅对冷却速率的控制依赖于规模和设备。规模和设备的变更将通过充分的风险分析、确认和验证,确保冷却速率(CPP)能够交付满足质量标准的原料药。
起始物料:CP-6和CP-8。这些测试要保留,同时用于确定新的生产/供应商。
重要物料属性:见第二篇起始物料选择中对中风险与低风险的论证。
表-2.3.S.2.6-14 重要物料质量标准及论证
|
重要物料属性 |
质量标准 |
论证 |
|
CP-6: | ||
|
CP-4 |
≤0.3% |
确保在原料药中,CP-4(≤0.3%) CP-5和CP-3(各≤0.1%)的总量≤10 ppm |
|
CP-5 |
≤0.1% | |
|
CP-3 |
≤0.1% | |
|
CP-8: | ||
|
CP-8-25I |
≤0.05% |
确保在原料药中≤0.10% |
|
CP-8-24I |
≤0.05% |
确保在原料药中≤0.10% |
图-2.3.S.2.6-25对整个设计空间的内识别的CPP和CQA(IMA)进行了总结:

图-2.3.S.2.6-25 步骤1和步骤2识别的CPP和CQA(IMA)
2.3.S.2.6总结
- 基于对Sakuramil 制剂产品QTPP的理解,以及对影响制剂开发的物理、化学、生物和微生物属性的知识和理解,确定了潜在的Sakuramil原料药关键质量属性。
- 从安全、环境、法规、经济、控制、产能(简称SELECT原则)角度,经过迭代,开发出具有成本效益、安全可控、环境友好的Sakuramil原料药商业化制造工艺。
- 基于商业可得性、与API结构相似性、合成步骤数和对起始物料的控制策略,对起始物料CP-6及CP-8的选择进行了论证。
- 通过开发和规模放大生产工艺获得的知识,以及对化学和制造工艺的机理和动力学理解,将工艺划分为若干单元操作,通过因果矩阵风险管理工具,对识别出四个目标区域(Focus Area):步骤1-反应,步骤1-结晶,步骤2-反应,步骤2-结晶。
- 进行中心复合设计与27-3部分因子设计,确定每个目标区域的工艺参数(考虑规模与设备)和物料属性与关键质量属性的函数(具有统计学和实际意义上显著)关系,识别出关键工艺参数及重要物料属性,最终输出整体制造工艺的设计空间和控制策略。
声明:本文转载来源于公众号【注册圈】,文章版权归原作者所有,如若了解更多信息可自行前往查看。
原文链接:https://mp.weixin.qq.com/s/hyoUE-dIG2qyonWn9LNbIQ声明:本文转载来源于公众号【注册圈】,文章版权归原作者所有,如若了解更多信息可自行前往查看。
原文链接:https://mp.weixin.qq.com/s/hyoUE-dIG2qyonWn9LNbIQ
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com