追根溯源教育产生于人类的什么过程中(追根溯源)
对于骨痛、肢体无力的患者该如何诊断和鉴别诊断?骨活检提示Paget骨病对诊断有何指导意义?一起学习一下最新一期(11月19日)Neurology杂志的临床推理病例吧!
医脉通编译整理,未经授权请勿转载。
病史及辅助检查
患者为56岁男性,因“上感后肢体无力和疲劳加重”就诊。1年前,患者渐感无力,最初表现为双臂举高困难;6个月前开始逐渐出现行走困难(坐位时难以起立和爬楼梯困难)。此外,约1.5年前开始出现逐渐加重的骨骼和关节疼痛。
患者吸烟,自18岁起每天吸烟1/2包。既往病史有慢性阻塞性肺疾病。否认近期疫苗接种史。无明显神经系统疾病家族史。父亲56岁死于心脏病,母亲死于早发性阿尔茨海默病。患者发育正常。
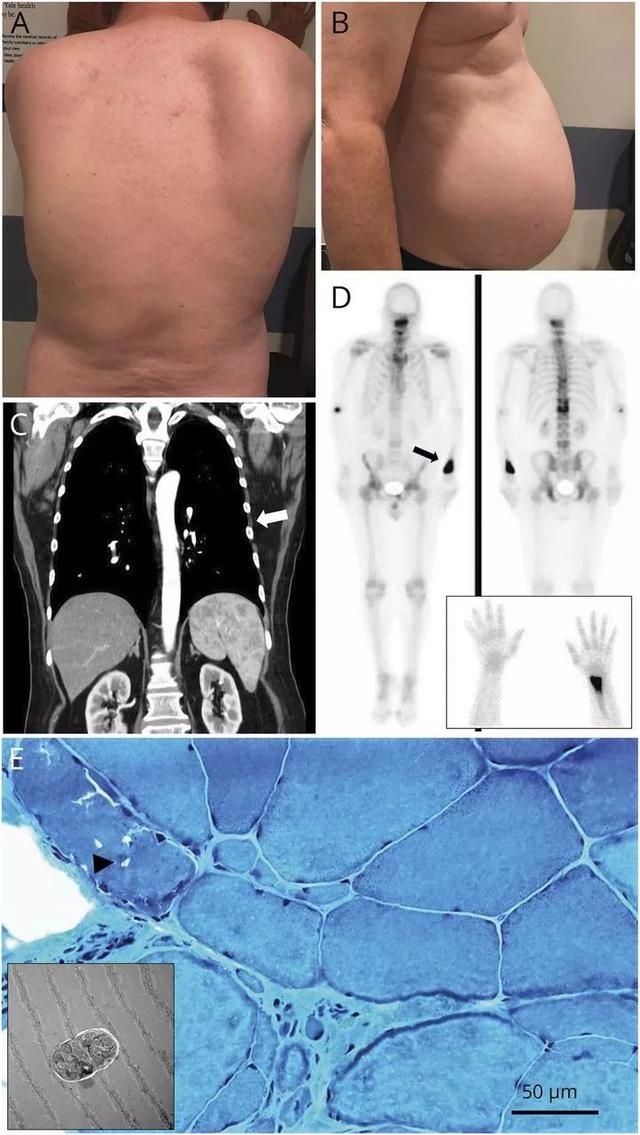
考虑到患者在急性呼吸系统疾病背景下出现无力恶化,最初怀疑是慢性炎症性脱髓鞘性多发性神经根神经病(CIDP)急性发作。但其无感觉症状且反射完整,因此在考虑腰穿之前先进行了脊柱MRI检查,结果显示除了C4和C7椎骨硬化病变外无明显异常。头颅MRI也正常。上述椎骨病灶骨扫描显示颈椎、胸椎和腰椎存在数个病灶活动度增加,左侧桡骨不均匀性硬化性和溶解性病灶(图D)。
图 关键临床特征和诊断结果。(A)右侧肩胛骨外翻;(B)腹部突出;(C)胸部CT扫描可见胸壁肌肉萎缩(白色箭头);(D)骨扫描显示左侧桡骨远端存在不均匀的硬化和溶解性病灶,颈椎和胸椎可见数个病灶活动增加;(E)Gomori三色染色显示纤维大小不一,可见镶边空泡(黑色箭头),插图显示带有包涵物的镶边空泡。
实验室检查显示碱性磷酸酶轻度升高(
135IU/L,正常40-115 U/L),而肌酐激酶(CK)(121 U/L,正常44–196U/L)、醛缩酶(4.8 U/L,正常1.0–7.5U/L)和血清钙(9.7 mg/dL,正常8.6–10.3mg/dL)正常。炎症性指标、风湿性指标、内分泌相关指标均未见明显异常。血清κ和λ轻链轻度升高,但血清蛋白电泳和免疫固定正常。
鉴于多处骨病和长期吸烟史,需考虑转移性病变,但胸部、腹部和骨盆CT扫描未发现隐匿性恶性肿瘤的证据。胸部CT显示胸壁肌肉脂肪萎缩(图C)。PET扫描仅显示椎骨病灶摄取增加。患者诊断不明并于4个月后转诊至神经肌肉诊所。在此期间,其无力继续发展,并且开始频繁跌倒。
问题思考:
1. 患者的骨病灶与其无力之间是否有联系?
2. 鉴别诊断有哪些?
鉴别诊断
查体可见右侧肩胛骨外翻(图A),肩带肌肉轻度萎缩和腹部突出(图B)。无力近端>远端(三角肌:3/5;髂腰肌:3 /5;二头肌、三头肌、腕屈、手指伸肌、股四头肌和脚踝背屈:4至4 /5)。握力降低(右侧16公斤,左侧17公斤,正常范围28-59公斤)。颅神经、反射和感觉未见明显异常。无小脑体征,可见步态蹒跚。
多发性硬化性骨病伴无力使得需考虑副蛋白血症,如多发性骨髓瘤和POEMS综合征或转移性骨病[1,2]。根据初步检查,尚无确凿证据支持这些病因。左侧桡骨病灶活检显示Paget骨病(PDB)。
对于近端无力的鉴别很多,包括肌病和非肌病性病因。考虑到无力的近端分布,应高度考虑炎症性肌病、皮肌炎(DM)和多发性肌炎,但肌酶正常、自身抗体阴性且无任何典型的DM皮肤损伤使得这些诊断可能性不大。包涵体肌炎(IBM)也应考虑,但该病股四头肌一般会比髋屈肌无力更明显,并且远端肌肉会早期受累,如手指屈肌和脚踝背屈肌。因患者肩带肌无力和肩胛骨外翻需考虑遗传性肌病,如四肢带状肌营养不良和面肩肱型肌营养不良。但其无面部受累和腿部早期受累对于后者来说相对不典型,而且两者通常早期起病。此外,非肌病性原因也需考虑。Lambert-Eaton重症肌无力综合征可能表现为近端无力,但患者缺乏易疲劳性或自主神经症状。无痛进行性无力也需要考虑运动神经元病,但其无上下运动神经元体征。如前所述,反射完整和缺乏感觉症状使得CIDP不太可能。但这些情况均不能解释PDB。
为了评估患者无力的病因,患者接受了肌电图检查。结果显示三角肌波幅降低,轻度多相运动单位动作电位;三角肌、二头肌、髂腰肌和股外侧肌可见早期募集;无自发性活动或提示肌膜激惹的证据(通常见于炎症性肌病)。
上述表现提示肌病,为此患者接受了右侧股四头肌活检,结果显示肌纤维大小不一和中央核增加,也可见少量镶边空泡。电子显微镜(EM)显示空泡中有内含物(图E)。
问题思考:
1. 哪些临床疾病与患者的临床表现相符?
进一步鉴别诊断
镶边空泡是许多肌病的非特异性表现,包括散发性IBM(sIBM)和肌原纤维肌病(MFM)。如前所述,患者的肌肉受累模式在sIBM中并不常见。肌肉组织学上未见无定形或粒状物质,并且EM未显示出肌原纤维变性以支持MFM。有趣的是,在伴包涵体肌病、Paget's骨病及额颞叶痴呆(IBMPFD)中也可见镶边空泡。鉴于患者近期被诊断为PDB,因此需高度怀疑IBMPFD [3-5]。
IBMPFD是一种独特的临床综合征,具有不同的临床表现[3]。在80%-90%的患者中发现肌病,而30%的患者表现为孤立性肌病。尽管无力通常发生在近端,但可能会累及远端肌群。血清CK通常正常或轻度升高,肌电图为肌源性改变(偶尔也有神经源性改变的报道)。肌肉活检通常显示出非特异性变化,包括萎缩、肌纤维大小不一、镶边空泡以及内含含缬酪肽蛋白(VCP)、泛素或TDP-43。
大约50%的患者会发展为PDB,通常比典型表现早十年,而近30%的IBMPFD患者会发展为额颞叶痴呆(FTD),通常为行为变异型。后者通常见于疾病后期,因此患者应接受神经心理学检测。该患者神经认知测试显示非遗忘性轻度认知障碍,缺损的模式提示额叶和顶叶受累,执行功能有轻度损害。
IBMPFD的三联征仅见于12%的患者,包括肝脂肪变性、白内障、扩张型心肌病、括约肌功能紊乱、感觉运动性轴突神经病和小脑体征等在内的其他特征也有报道[4]。
问题思考:
1. 如何确诊?
确诊
IBMPFD是一种常染色体显性遗传性疾病,继发于9p13.3染色体上VCP基因突变[3]。遗传检测对于确诊至关重要。该患者VCP基因第5外显子有致病性G>A错义突变,导致精氨酸取代组氨酸,从而得以确诊。
VCP参与DNA修复、凋亡、细胞周期控制和蛋白质降解。研究显示大约45个不同的突变与IBMPFD有关[5]。这些突变可能会破坏VCP在蛋白质稳态中的正常作用,从而导致异常蛋白聚集[6]。
最近研究显示非VCP蛋白(SQSTM1、HNRPNA2B1和HNRNPA1)的突变也与IBMPFD有关。随着研究深入,IBMPFD的临床表现也不止经典的三联症,且存在其他基因突变,因此有学者提出将IBMPFD定义为多系统蛋白病[7]。该术语强调了多种系统受累和病理的相同性,同时允许遗传和表型的异质性。
讨论
IBMPFD是一种罕见的多系统疾病,因其表现多异,常被误诊。没有治疗可以改变或阻止其进展。理疗、职业治疗以及辅助设备可以促进功能恢复。尽管没有令人信服的证据表明治疗改变疾病进展,但双膦酸盐可能会改善PDB患者的骨痛[8]。选择性5-羟色胺再摄取抑制剂可能对FTD患者的行为症状有益[9]。疾病后期,患者可能会出现呼吸或心脏衰竭。
该病例展示了IBMPFD诊断的难点。对这种异质性多系统疾病做出诊断可能并不容易。在评估患有肌病和骨病的患者时,需纳入鉴别诊断。早期识别可能有助于更好地管理相关的并发症并改善生活质量。
参考文献:
[1] Batsis JA, McDonald FS. 76-year-old man with backpain and progressive leg weakness. Mayo Clin Proc 2005;80:259–262.
[2] Waning DL, Guise TA. Molecular mechanisms of bonemetastasis and associated muscle weakness. Clin Cancer Res 2014;20:3071–3077.
[3] Kimonis VE, Mehta SG, Fulchiero EC, et al. Clinicalstudies in familial VCP myopathy associated with Paget disease of bone andfrontotemporal dementia. Am JMed Genet A 2008;
146A:745–757.
[4] Weihl CC, Pestronk A, Kimonis VE. Valosin-containingprotein disease: inclusion body myopathy with Paget’s disease of the bone andfronto-temporal dementia. Neuromuscul Disord 2009;19:308–315.
[5] Al-Obeidi E, Al-Tahan S, Surampalli A, et al.Genotype-phenotype study in patients with valosin-containing protein mutationsassociated with multisystem proteinopathy. Clin Genet 2018;93:119–125.
[6] Watts GD, Wymer J, Kovach MJ, et al. Inclusion bodymyopathy associated with Paget disease of bone and frontotemporal dementia iscaused by mutant valosin-containing protein. Nat Genet 2004;36:377–381.
[7] Taylor JP. Multisystem proteinopathy: intersectinggenetics in muscle, bone, and brain degeneration. Neurology 2015;85:658–660.
[8] Wat WZ. Current perspectives on bisphosphonatetreatment in Paget’s disease of bone. Ther Clin Risk Manag 2014;10:977–983.
[9] Young JJ, LavakumarM,Tampi D, Balachandran S,TampiRR. Frontotemporal dementia: latest evidence and clinical implications. TherAdv Psychopharmacol 2018;8:33–48.
原文索引:TaraTorabi, Anita Huttner, Richard J. Nowak and Bhaskar Roy. Clinical Reasoning:Progressive pro
,ximal weakness in a 56-year-old man with bone pain. NeurologyNovember 19, 2019 93:939-944. http://n.neurology.org/content/93/21/939.extract.

免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






