节点电压公式详解(Angew钠电加点钾有奇效)

第一作者:Cheng zheng
通讯作者:Nana Wang
单位:University of Wollongong
【背景】
钠离子电池(SIBs)作为有前途的储能设备之一,在过去几年中引起了广泛的兴趣,因为钠的成本低,操作原理与锂离子电池(LIBs)相似,以及Na 离子的独特物理化学特性。在众多的候选阳极材料中,锡(Sn)特别有吸引力,因为它具有高理论容量(847 mAh g-1)、大体积密度(7.28 g cm-3)和低工作电压(0.3-0.4 V)。然而,Na 的大离子半径和重离子质量不仅在循环时引起巨大的体积变化(~420%),而且还延迟了电荷在体积中的传输。因此,固体电解质界面(SEI)的反复形成、不可控的粒子粉碎以及循环过程中的电极开裂/脱落,导致低库仑效率和快速的容量衰减。为了解决这些问题,人们开发了多种策略,如尺寸控制到纳米,与碳材料的均匀复合,截止电压的精细控制,等等。例如,广泛报道的Sn/C纳米复合材料总是涉及费力的制备、复杂的仪器和昂贵的试剂。此外,纳米复合材料中的碳材料不仅降低了比容量,而且还降低了攻丝密度和初始库伦效率。因此,有必要寻求新的方法来改进Sn阳极。使用微小的锡颗粒(μ-Sn)作为阳极材料可以有效地缓解对纳米材料的制备成本、压实密度和初始库仑效率的担忧。但如何保持其稳定的循环是社会的一个巨大挑战。到目前为止,针对μ-Sn设计的策略还很少报道。
【工作介绍】
本工作将K 离子引入到醚基电解质中。然后,K 和Na 离子被电场驱动到电极表面。但是K 离子不会被还原或纳入μSn中,因为Nernst方程、分子动力学(MD)模拟和密度泛函理论(DFT)计算估计的氧化还原电位低,解离能量大。K 离子会聚集在 "热点 "上,并通过静电屏蔽作用减缓局部的钠化,促进均匀的电化学反应。它增强了电极的稳定性,正如化学机械模型和扫描电子显微镜(SEM)图像所证明的。因此,该电极在2 A g-1的条件下经过3000次循环后保持了565 mAh g-1的高比容量,比电解质中没有K 的情况好很多。在全电池和μ-Bi中也证明了K 离子的改进性能。类似的概念已经应用于金属锂阳极,但据我们所知,还没有应用于合金型阳极。更重要的是,这种方法不涉及费力的制备、复杂的仪器和昂贵的试剂,因此显示出对其他SIB阳极材料的潜力。
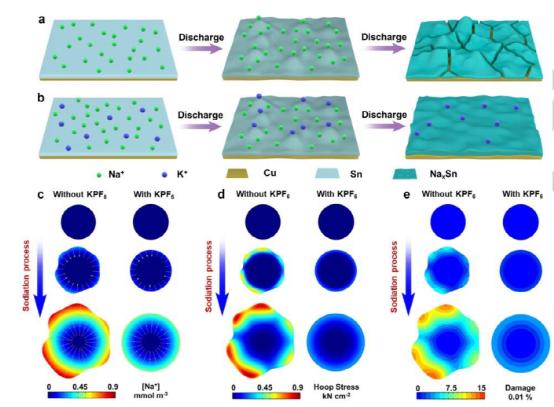
图1、K 离子的静电屏蔽作用,增强了电极的稳定性。(a, b) 合金型阳极(a)没有或(b)在电解质中加入K 的结构演变。(c)Na浓度、(d)环向应力和(e)在没有或有电解液中的K 的情况下,在钠化过程中对Sn的结构破坏的化学-机械模型。
图1说明了K 对增强电化学性能的静电屏蔽作用。由于活性材料、聚合物粘合剂和导电碳在电极中的不均匀分布,Na 在特定部位的局部富集和优先钠化几乎是不可避免的(图1a)。局部钠化的进展将引起内在应力的不均匀分布,并最终导致电极破裂。相比之下,添加K 将有效地缓解上述负面影响。如上所述,在这种情况下,K 比Na 更难被还原。因此,当K 被电场驱动到活性材料附近时,它不会被还原,而是优先吸附在Na 离子应该占据的热点处(图1b)。
K 的持续积累将呈现出一个内在的电场,并抵消了Na 到达这些位置的驱动力,这被称为典型的静电屏蔽。然后,在热点位置的局部钠化将放缓,有利于均匀的体积膨胀并改善电极的稳定性。这个结论也得到了环向应力和损伤分析的支持。为了简化计算,只有活性材料(μ-Sn),而不是整个电极,被考虑到模拟中,因为它们在电极中占主导地位。此外,它们的体积变化是结构应力的主要来源。
尽管μ-Sn经历了复杂的相变,但在这些相变过程中结构应力的演变是相似的。因此,为了清楚起见,本化学机械模型只使用了其中一个相变,即Sn/NaSn3。图1c-1e显示了在这一转变过程中Na浓度、环向应力和μ-Sn的结构损伤的演变。在一个典型的过程中,由于内部体积膨胀,钠的外壳逐渐向外挤压,导致粒子表面产生拉伸箍应力。拉伸应力随着钠化过程而逐渐累积。当拉应力超过NaxSn的阈值时,颗粒会变形或断裂以释放应力。在没有K 的情况下,由于各向异性的形态、粗糙的颗粒表面和电极中不均匀的材料分布,Na 通量容易诱发不均匀的钠化过程和μ-Sn的各向异性膨胀(图1c)。然后,表面的拉应力加剧,增加了潜在的粒子损伤(图1d和1e)。相反,K 通过静电屏蔽使粒子表面的Na 分布正常化,促进了均匀膨胀(图1c)。然后,相邻材料网格的拉应力相互抵消,导致较低的拉应力和较少的颗粒损坏(图1d和1e)。这些结果支持K 的加入有可能促进均匀的钠化和减少颗粒的损伤。

图2、通过KPF6改善电极的稳定性和机械性能。(a-f) 电解液中没有K 或(d-f)30个周期后的电极的SEM图像。(g, j) AFM图像(插图)及其相应的高度曲线,(h, k) 力的反应及其相应的卡通插图(插图),(i, l) 分别在没有或在电解质中加入K 后的电极的杨氏模量分布。
在没有K 的电解液中循环30次后,在电极表面很容易看到裂缝和断裂(图2a和2b),表明循环后的电极稳定性较差。同时,电极材料变得松散并从集流体上脱落(图2c)。结果表明,巨大的应力对电极造成了明显的损害,这增加了电荷传输电阻,导致容量下降。相反,在电解液中含有K 的情况下,基本上保持了电极的完整性(图2d和2e)。电极表面没有明显的裂缝。电极材料仍然保持致密并牢固地附着在集流体上(图2f)。这些结果证实了电极稳定性的增强,与上述理论预测的结果非常吻合。
AFM得出类似的结论。图2g显示了在电解液中没有K 的情况下,3个循环后电极的高度曲线和表面形貌,电极表面是粗糙的。表面上的台阶和/或山脊会强化电场,丰富局部的Na 浓度,从而加速局部生长,增加应变/应力。然后,电极材料变形,在反复循环后出现裂纹。而在电解液中含有K 的电极循环是相对光滑的(图2j),与循环前的情况相似。这种良好的稳定性也可能与该电极对结构应力的反应有关。如图2h所示,在没有K 的情况下循环的电极的力的曲线显示了电极破裂的典型特征,即力随着压痕深度的增加而下降。它符合SEM图像所观察到的情况。然而,这种独特的特征并没有出现在电解液中含有K 的电极循环的力的曲线上(图2k),这表明在这种情况下对力有良好的抵抗力。这方面的见解可能与μ-Sn的均匀膨胀有关,它使内在应力在一定程度上被抵消。因此,电极对本征应力的耐受性更好,并表现出卓越的稳定性。这一结论直接得到了杨氏模量的支持。无K 循环的电极的杨氏模量只有2.7GPa,比有K 的8.3GPa小很多(图2i和2l)。

图3、K 离子不参与μ-Sn的合金化/焊接反应。(a) 电解液中含有或不含有K 离子的电极循环的CV曲线,(b) EDS光谱和(c) XPS光谱。(d) K /K和K /KxSn的氧化还原电位随K 离子浓度的变化。(e) Li /Li和Li /LixSn的氧化还原电位随Li 离子浓度的变化。(f) 电解液中有无Li 的Sn||Na的循环性能,(g, h, i) 电解液中Na 和K 的典型溶解结构快照,(j) RDF曲线,(k) LUMO的能级,(l) Na -2diglyme和K -3diglyme的溶剂化能量。(k)中的插图显示了分子轨道的等值面,(l)中的插图描述了相应的溶剂化结构。
K 对电化学性能的另一个可能影响是在循环过程中与μ-Sn合金化。为了澄清这一点,在循环的电极上测量了CV曲线、LSV(线性扫描伏安法)曲线、能量色散光谱(EDS)和X射线光电子光谱(XPS)。如图3a所示,在电解质中含有或不含有K 的电极循环显示出相同的阴/阳极峰,表明K 对电化学反应的影响可忽略不计。换句话说,K 不参与合金化/去合金反应。这一结论也被EDS和XPS所证实。图3b显示了在电解液中含有或不含有K 的完全放电电极的EDS光谱。无论是否加入K ,在这两个电极中都没有检测到K元素在3.31 keV (Kα)和3.59 keV (Kβ)的明显信号。这些结果排除了K参与合金化/去合金反应的可能性。同时,在用K 循环的电极中Na/Sn的原子比是~3.84,接近Na15Sn4的数据(3.75),该数据被公认为是Sn的完全放电产物。这一结果表明,K 提高了电极中Sn的利用率,因为它减少了Na 在热点上的吸附,使Na 可以在其他位置上进行电化学合金化。电极中没有K 也被不同深度的XPS光谱所证明(图3c),没有观察到K的可见峰。K 的加入并不直接改变电极表面的固体-电解质-中界面(SEI)膜。因此,在两种情况下,Rct的差异很小。这一结果也得到了LSV的支持。电解液中含有或不含有K 的电池显示出类似的曲线,证实了K 物种没有被还原。
为了在理论上理解这一结果,详细讨论K /K(Na /Na)的实际氧化还原电位和我们案例中K /KxSn的合金化电位是非常必要的。众所周知,K /K的标准氧化还原电位(-2.931 V vs. SHE)低于Na /K的氧化还原电位。K /K的实际氧化还原电位会随着K 的低浓度而进一步降低(蓝线,图3d)。在本例中,K 的浓度为0.02M,导致氧化还原电位为-3.031V( vs. SHE),使得K /K的电化学还原更加困难。然而,K 有可能与Sn进行电化学合金化,在~0.32 V(vs. K /K)开始。如果参考电极被归一化为SHE,K /KxSn的起始电位将是-2.611 V(vs. SHE)。然后,在本案例中,低浓度被考虑在内。K /KxSn的氧化还原电位被估计为-2.711 V(vs. SHE,红线,图3d),几乎与Na /Na的相同。考虑到电极过电位,K /KxSn的氧化还原反应必须发生在这个电位以下。这意味着,如果截止电压被控制在>0.01V(vs. Na /Na),KxSn的形成将被抑制。这些结果解释了为什么K 不参与SIBs中Sn的电化学反应。然而,如果把K 换成Li ,情况就完全不同了。因为Li /LixSn的起始电位是-2.44V(vs. SHE,图3e),Li 有可能在钠化过程中与Sn合金化。这将使有限的Li 的消耗,从而使电化学性能不能与K 相比(图3f)。因此,选择一个合适的阳离子进行静电屏蔽是至关重要的。
基于Nernst方程的讨论是简化的,因为它没有考虑到溶剂化结构。因此,进行了分子动力学(MD)模拟以反映电解质中的情况。如图3g和3i所示,电解质中的Na 通过Na···Odiglyme与两个DME分子相互作用,而K 通过K···Odiglyme与三个DME分子配位。径向分布函数(RDFs)表明,Na比K更接近Odiglyme(图3j),但Na的配位数(CN)比K的小。这可能是由于K 尺寸比较大。然后,取溶剂化结构来计算最高占有分子轨道(HOMO)和最低未占有分子轨道(LUMO)的能级。通常情况下,LUMO的能量越低,表明该物种的优先还原。[K(diglyme)3] 的LUMO位于-1.79 eV(图3k),高于[Na(diglyme)2] (-2.14 eV)。所以,[Na(diglyme)2] 将率先被还原。这一结果再次证实了K 在电解质中相对较好的电化学稳定性。除了热力学,动力学也阻碍了K 在反应中的参与。因为溶剂化结构在还原前需要解离,K 和Na 的溶剂化能可以被认为是评估能量屏障的一个指标。如图3l所示,[K(diglyme)3] 显示出比[Na(diglyme)2] (-4.69 eV)更大的溶剂化能(-24.97 eV)。这些数据意味着[K(diglyme)3] 的解离需要比[Na(diglyme)2] 的解离更多能量。换句话说,[K(diglyme)3] 比[Na(diglyme)2] 更稳定。这一结论也得到了静电势(ESP)图的支持。Na(diglyme)2] 的平均ESP比[K(diglyme)3] 的更正,这使得它对电子攻击高度活跃,有利于电化学还原。简而言之,高配位数、大的溶剂化能、高的LUMO能级和离域电荷分布使得K 在电解质中更加稳定,这与马库斯理论的预测非常一致。基于这一理论,具有较少溶剂分子和局部电荷分布的巨型结构具有小的重组能垒。在这种情况下,溶剂化的Na 离子比K 离子的对应物更活跃。所有这些结果在理论上和实验上都证明,K 不参与Sn的电化学反应。

图4、KPF6增强的电化学性能。

图5、NVP@C||Sn和Bi||Na的电化学性能。
总之,K 离子被引入电解质以促进均匀的反应和电极的稳定性,这在化学机械模型、SEM和AFM中得到了证明。CV曲线、EDS和XPS光谱表明,K 离子不直接参与电化学反应,这一点得到了由Nernst方程估计的氧化还原电位、LUMO的能级和基于MD模拟得出的溶剂化成分的解离能的支持。
静电屏蔽的作用使μ-Sn在2 A g-1的条件下经过3000次循环后能提供565 mAh g-1的容量。相比之下,在电解液中没有K 的情况下循环的电极只能存活34次。K 带来的益处在高负荷电极、全电池和其他阳极材料中也得到了证实,反映了未来的巨大潜力。这种策略特别耐人寻味,因为不需要费力的准备、复杂的仪器和昂贵的试剂。
Electrostatic Shielding Boosts Electrochemical Performance of Alloy-Type Anode Materials of Sodium-Ion Batteries
Angewandte Chemie International Edition ( IF 16.823 ) Pub Date : 2022-11-30 , DOI: 10.1002/anie.202214258
Cheng zheng,Deluo Ji,Qian Yao,Zhongchao Bai,Yansong Zhu,Chuanhao Nie,Duo Liu,Nana Wang,Jian Yang,Shixue Dou
,免责声明:本文仅代表文章作者的个人观点,与本站无关。其原创性、真实性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容文字的真实性、完整性和原创性本站不作任何保证或承诺,请读者仅作参考,并自行核实相关内容。文章投诉邮箱:anhduc.ph@yahoo.com






